加氫催化劑活性相形貌結構認識與研究進展
2023-03-14 12:02:00李晨曦李會峰鄭愛國楊清河
石油煉制與化工 2023年2期
李晨曦,張 樂,李會峰,鄭愛國,楊清河
(中石化石油化工科學研究院有限公司,北京 100083)
隨著能源結構向低碳方向轉型,油品質量標準也不斷升級。同時,世界原油劣質化、重質化程度加劇,為生產符合質量標準的清潔油品,亟需進一步提升加氫催化劑的催化性能(選擇性、活性和穩定性)。例如,柴油加氫精制過程中,隨著脫硫深度的增加,加氫催化劑的失活加快,因此,新研發的加氫催化劑面臨著兼具高活性和高穩定性的嚴峻挑戰[1-3]。傳統負載型加氫催化劑常用的活性金屬組元組合為Co Mo,Ni Mo,NiW,而開發加氫催化劑的核心是活性相結構的設計[4]。迄今為止,研究者提出的關于加氫催化劑活性相結構的理論模型有十余種,其中Tops?e等[5]提出的Co-Mo-S活性相結構模型已得到研究者的廣泛認可。在過去的幾十年中,隨著先進分析表征技術,如原位穆斯堡爾譜、X射線吸收精細結構(EXAFS)、球差校正掃描透射電子顯微鏡(Cs-STEM)、掃描隧道電子顯微鏡(STM)以及分子模擬技術等的發展,使學者們可以更直觀地觀測和剖析活性相的結構信息,為進一步深入認識加氫催化劑活性相的結構特點提供了新的技術支撐。
本文從加氫催化劑活性相形貌結構的表征和認識、影響活性相形貌結構的因素以及活性相形貌結構與催化劑性能之間的關系等方面進行歸納總結,以期為后續新型加氫催化劑的高效研發提供理論依據。
1 活性相形貌結構的認識
丹麥Aarhus大學和Tops?e團隊[6-7]利用STM方法研究了Au基底上 MoS2、Co(Ni)-Mo-S的圖像(見圖1),發現單獨的MoS2表現為規則的三角形形貌,助劑Co在MoS2的(010)S-edge上修飾形成六邊形形貌的Co-Mo-S相。此外,研究者還發現助劑Ni對MoS2的修飾作用與助劑Co并不完全相同,除了修飾S-edge形成Type A Ni-Mo-S相外,還會在(100)Mo-edge和S-edge交叉處形成切角,構成新的(110)邊位,形成有12個邊位的Type B Ni-Mo-S相,導致Co-Mo-S和Ni-Mo-S在催化活性和選擇性上的差異。Tops?e團隊[8]又利用配備高角度環形暗場探測器-高分辨率掃描透射電子顯微鏡(HR HAADF-STEM)觀察負載在石墨上的 MoS2、Co(Ni)-Mo-S的形貌結構特點(見圖2),無助劑修飾的MoS2表現為圓角三角形,助劑Co、Ni的引入會促進MoS2的形貌由圓角三角形逐漸轉變為圓角六邊形,暴露出(110)邊位。
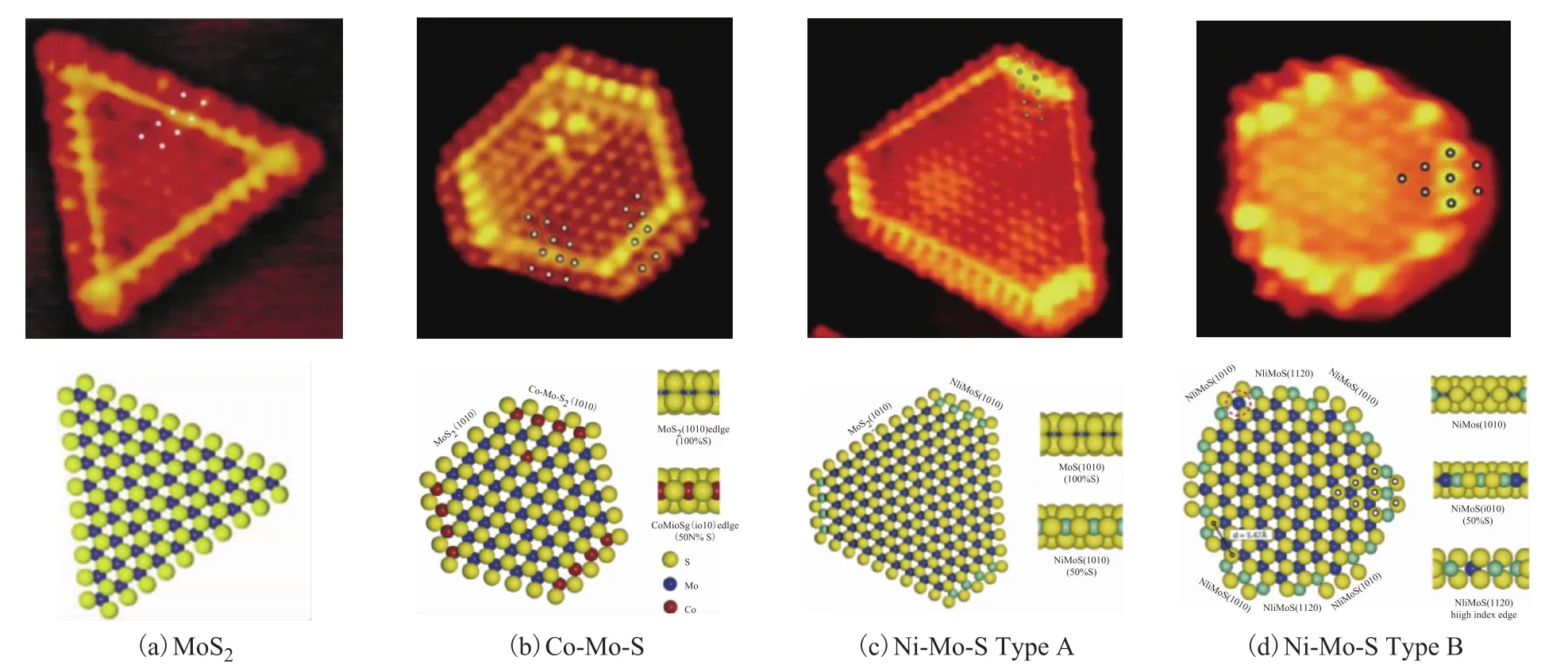
圖1 負載在Au載體上的金屬硫化物的STM圖像及其球棍模型[6-7]

圖2 負載在石墨載體上的金屬硫化物的HR HAADF-STEM 圖像[8]
工業加氫催化劑多以Al2O3為載體,由于Al2O3表面結構性質復雜,導致其負載的 MoS2、Co(Ni)-Mo(W)-S形貌結構與負載在 Au或石墨載體上時有較大差異。由于金屬與載體之間的相互作用(MSI)的不同,導致形成的Co-Mo-S活性相的形貌結構特點有一定差異,可分為TypeⅠ類相和TypeⅡ類相。TypeⅠ類相與載體之間通過Mo—O—Al鍵結合,MSI較強,金屬硫化不完全,其形態為單層片晶;而TypeⅡ類相與載體之間通過范德華力結合,MSI較弱,金屬屬于完全硫化結構,其形態通常為多層片晶[9]。Baubet等[10]以δ-Al2O3為載體,制備負載8%(w,下同)MoO3的Mo/Al2O3催化劑,在100%H2S、550℃的條件下硫化后,通過HR HAADF-STEM觀察到催化劑表面同時存在三角形和六邊形的MoS2活性相;此外,還能清楚地觀察到許多尺寸小于1 nm的無定形金屬團簇分散在載體表面。此前,相關學者的研究[11]表明,MoS2團簇具有強烈的結構-尺寸效應,其鍵能以及形成硫空位的趨勢會隨尺寸變化。其中尺寸小于2 nm的MoS2團簇的S/Mo比例增加會導致其內部和邊緣結構能量發生明顯變化,從而可能會改變其結構性質。何文會等[12]使用Cs-STEM觀察不同硫化程度的5% WO3/Al2O3催化劑時發現,除了清晰可見的活性相片晶結構以外,載體表面還存在很多形貌結構不規則的金屬團簇。該研究團隊根據原子間距和形貌結構對活性金屬的存在形式進行了分類:將尺寸大于2 nm、形貌結構接近三角形或六邊形的活性相命名為片晶,而尺寸小于2 nm、且形貌結構不規則的命名為金屬簇,單個原子即為單原子(見圖3)。隨著活性金屬的硫化程度提高,單原子、金屬簇形貌結構在載體表面所占比例逐漸減少,但并未完全消失。另外,借助Cs-STEM發現商用、新鮮硫化的NiMo/γ-Al2O3催化劑(22.0% MoO3,4.0% NiO)中也存在單原子和金屬簇[13]。Eijsbouts等[14]發現,不同金屬含量(2.0% MoO3和6.5% MoO3,Co/Mo原子比為0.36)的Co Mo/Al2O3催化劑中均存在金屬簇,結合能量色散X射線光譜儀(EDX)分析,局部Mo/Al原子比小于0.1的區域中具有更多的金屬簇,隨著Mo/Al原子比增加,載體表面被更大的MoS2活性相覆蓋。
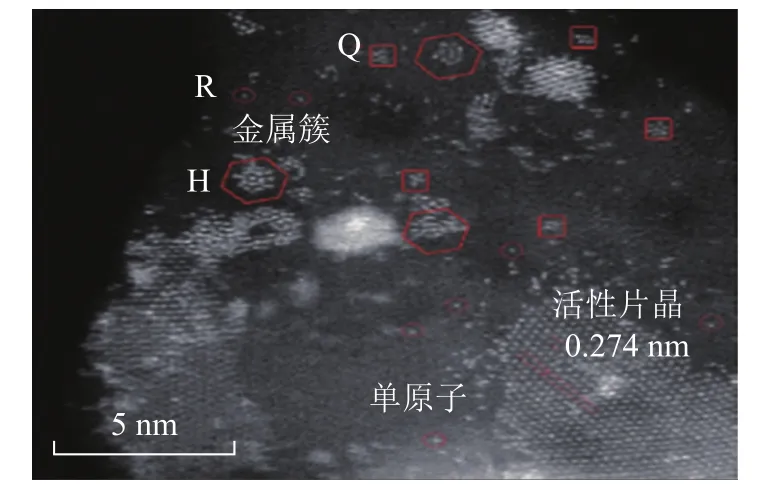
圖3 催化劑不同形貌結構活性相分類[12]
從以上研究結果可以看出,通過TEM以及Cs-STEM等表征手段可以清楚直觀地觀測催化劑的活性相形貌結構。不同表征手段對活性相形貌結構的認識維度不同:借助TEM能清楚地觀察大尺寸活性相(例如片晶)的形貌結構特點,統計片晶的尺寸和層數分布;而HR HAADF-STEM能直觀地觀察到更精細的活性相結構,有助于區分不同形貌活性相的邊緣活性位點分布情況。另外,由于MSI對催化劑活性相形貌結構有顯著影響,對于以Al2O3為載體的工業催化劑,其活性相形貌結構更加復雜,表現為單原子、金屬簇和片晶多種形貌結構共存。
2 影響活性相形貌結構的因素
加氫催化劑由載體和負載于其上的活性金屬組成,Al2O3由于其較好的孔結構參數和表面結構性質,常用作加氫催化劑的載體材料。因此,從催化劑制備角度來講,催化劑的活性相形貌結構主要與金屬前軀物分子結構、載體的性質以及硫化條件緊密相關。目前已有很多研究者致力于改變金屬前軀物分子結構以提升催化劑的性能,例如,Li Huifeng等[15]發現常規技術制備的金屬前軀物溶液中僅含有鉬雜多陰離子[HPMo12O40]2-,而采用其發明技術制備的金屬前軀物溶液中則含有鈷鉬雜多陰離子[H2CoMo4PO16]-和[HCoMo6O2]2-,表明該發明技術促進了鈷鉬雜多化合物的定向生成,從而使催化劑的加氫脫硫(HDS)活性提高了10%~12%。Bara等[16]研究α-Al2O3不同晶面對MoS2生長方向以及尺寸的影響,通過HR HAADFSTEM發現,在MSI較弱的C(0001)晶面上,活性金屬以基面鍵合的方式分散在載體表面,更容易被硫化成尺寸較大的多層MoS2片晶。而R(102)晶面的MSI太強,導致活性金屬氧化物高度分散,難以被硫化。此外,Araki Y等[17]認為,硫化條件的差異也會反過來影響活性金屬物種在載體表面的分散狀態。因此,當金屬前軀物分子結構固定不變時,載體表面結構性質和硫化條件都會引起活性相形貌結構的變化。
2.1 載體性質
Al2O3的表面結構性質,如結晶度和表面羥基數量等對活性金屬在載體表面的存在形式影響較大,因此對載體表面性質進行修飾可以有效調控活性相形貌結構。目前常用的載體表面性質修飾方式可分為添加其他元素和不添加元素兩大類。
2.1.1 添加其他元素的載體改性
添加其他元素對載體改性的方法主要包括分別在Al2O3中引入Ti,F,P和絡合劑等,通過改變載體表面性質,從而影響MSI,引起活性相形貌結構變化。郭長友等[18]研究了TiO2修飾Al2O3載體表面對MoS2分散性的影響。TiO2優先占據 Mo-Al2O3強相互作用中心位置,從而顯著降低MSI,導致氧化態金屬分散性下降,MoO3晶粒長大,硫化后生成的MoS2片晶的平均尺寸和堆疊層數增加,說明MoS2片晶的形成過程受Al2O3表面性質控制,其尺寸與 MoO3晶粒大小有關。Guo Xingmei等[19]采用NH4F改性制備了一系列不同F含量的 NiWF(x)/γ-Al2O3催化劑,發現F能取代Al2O3表面的堿性羥基,減弱MSI,促進活性金屬硫化;但隨著F含量增加,催化劑的比表面積逐漸降低,活性金屬分散性變差,WS2片晶的平均尺寸和平均堆疊層數逐漸增加,對煤焦油的加氫脫氮(HDN)、HDS反應活性呈先增加后降低的趨勢。為了更好地控制活性相的生長程度,李會峰等[20]采用磷鎢酸作為鎢源引進P在含F的Al2O3載體上制備了PW/F-Al2O3催化劑,硫化過程中解離出的P能對WS2起到一定的隔離作用,有效防止活性金屬過度聚集長大,從而形成平均尺寸和層數適中的WS2片晶,表現出較高的4,6-二甲基二苯并噻吩(4,6-DMDBT)HDS活性。
此外,添加絡合劑也會對催化劑活性相產生影響,常用的絡合劑有檸檬酸(CA)[21]、乙二胺四乙酸(EDTA)[22]、十六烷基三甲基溴化銨(CTAB)[23]等。本文以常用的絡合劑CA為例。在催化劑制備過程中引入的CA主要發揮兩個作用:“修飾載體表面”有利于弱化MSI,“隔離活性相”有利于形成較小尺寸的活性相片晶。但這兩個作用所帶來的實際效果會隨著CA的引入方式、引入順序和添加量等的不同而發生變化[21]。Haandel等[24]在10%(φ)H2S/90%(φ)H2、2.0 MPa的硫化條件下,借助X射線光電子能譜(XPS)和EXAFS等表征方式,研究了不同硫化溫度下Co Mo催化劑中活性金屬存在形式的變化過程。研究發現,大部分的活性金屬(Co、Mo)在硫化溫度達到250℃時才能開始硫化;而添加CA后,大幅減弱了MSI,促使活性金屬在50℃時就開始硫化,生成的MoS2片晶平均長度和平均堆疊層數增加。Chen Jianjun等[25]將一定量的七鉬酸銨、硝酸鈷和不同比例的CA配制成混合液,以等體積浸漬法制備出Co Mo/Al2O3催化劑。相同硫化條件下,CA添加量越高的催化劑,其MSI越弱,生成的MoS2片晶平均長度變長和堆疊層數增高。通過CO吸附紅外光譜分析發現,其暴露出更多的S-edge,說明改變CA添加量會帶來MoS2形貌結構的變化。Zavala-Sanchez團隊[26]發現添加CA后,借助TEM統計得到的MoS2片晶的平均尺寸從(2.90±0.12)nm 降至(1.70±0.12)nm,平均堆疊層數從1.80±0.04增至2.20±0.05,同時提高了活性金屬分散性;此外,通過HR HAADF-STEM觀察到活性相形貌從三角形變為具有更高S-edge/M-edge(M為金屬)比例的六邊形。相比于TEM的統計結果,HR HAADF-STEM統計出的MoS2片晶的尺寸分布更均勻,易于區分單原子和金屬簇。該團隊繼續研究了CA對Co Mo/Al2O3催化劑活性相形貌結構的影響[27],同樣發現CA會大幅提高活性金屬分散性,使催化劑的活性相分散值從48%增至78%,出現更多分散較好的小金屬簇。但是添加CA反而使得活性相形貌從六邊形轉變為三角形,這與其對無助劑修飾的Mo/Al2O3催化劑的活性相形貌結構的影響恰好相反。此外,CA的引入方式對催化劑活性相的形貌結構也具有較大的影響。Takeshi等[28]使用后引入CA的方法,將CA配制成溶液,以等體積方法浸漬焙燒成型的Co Mo-B/Al2O3催化劑,經110℃下干燥制成催化劑。對比劑是將含有七鉬酸銨、硝酸鈷和CA的浸漬液以等體積浸漬法一次浸漬在B改性后的Al2O3載體上,以共浸漬法引入CA制備的催化劑。研究結果表明,引入CA的方式會影響活性金屬的硫化行為。通過原位EXAFS分析可知,后引入CA的方法有助于Mo金屬優先硫化或者促使Co、Mo兩種金屬同時硫化,避免Co金屬單獨硫化形成不活潑的硫化物(如Co9S8),從而更好地發揮Co的助劑修飾作用,形成更多的Co-Mo-S相,因此后引入法制備的催化劑對噻吩的HDS活性高于共浸漬法制備的催化劑。
2.1.2 不添加元素的載體改性
常用的不添加元素的載體改性方式主要指對載體進行焙燒和水熱處理。張軒等[29]將中國石化催化劑長嶺分公司生產的渣油降殘炭脫硫催化劑RCS-30的載體干條置于不同溫度下焙燒后,觀察其物性結構變化。隨著焙燒溫度從480℃逐漸升高至800℃時,焙燒成型的Al2O3載體的比表面積和表面羥基數量減少,MSI減弱,活性金屬聚集程度加劇,生成的MoS2片晶的平均長度和堆疊層數增加。Chen Wenbin等[30]發現,隨著擬薄水鋁石的焙燒溫度逐漸升高(500~900℃),生成的 γ-Al2O3的孔徑分布逐漸向大孔方向移動,平均孔徑增加;堿性羥基數目基本不變,酸性羥基數量減少,總羥基數量下降,使得生成的MoS2和Co-Mo-S活性相的占比減少;位于 MoS2片晶邊緣和角位的Mo原子的濃度逐漸減小,從0.43 mol/L降至0.28 mol/L,暴露出的反應活性位點減少。當焙燒溫度繼續升高,超過900℃時,Al2O3會發生晶型轉變[31]。Wang Xilong等[32]發現隨著載體焙燒溫度升高 (550,900,1 000 ℃),Al2O3晶型發生γ→δ→θ的轉變,同時載體的比表面積和孔道結構發生變化,Ni Mo/Al2O3催化劑表面活性 MoS2片晶的平均尺寸變長和堆疊層數增加。在不同空速下,催化劑NiMo/δ-Al2O3均表現出最高的二苯并噻吩(DBT)以及4,6-DMDBT脫硫活性,其中對4,6-DMDBT的HDS反應轉化頻率(TOF)約為催化劑NiMo/γ-Al2O3作用下的3.5倍。這與Laurenti等[33]關于 Mo/δ-Al2O3催化劑對4,6-DMDBT 的HDS活性研究結果是一致的,相同硫化條件下,負載在δ-Al2O3上的催化劑具有更高的HDS本征活性。
另外,水熱處理能調節Al2O3的晶粒大小以及載體表面的羥基分布情況。Li Huifeng等[34]將γ-Al2O3在180℃下水熱處理4 h后,于400℃下焙 燒 得 到 Al2O3-(γ-HT)。相 比 于 γ-Al2O3,Al2O3-(γ-HT)的晶粒尺寸從3.2 nm 增至6.6 nm。伴隨著晶粒尺寸的增長,載體的比表面積和孔體積明顯減小,表面羥基數目下降,活性金屬更容易被硫化,生成的(Ni)MoS2活性相的平均尺寸和平均堆疊層數隨著晶粒長大而顯著增加。張軒等[35]考察了水熱處理時間對渣油脫硫降殘炭催化劑RCS-30載體的影響。當水熱處理溫度為95℃時,隨著載體水熱處理時間從1 h延長到8 h,載體的平均孔徑逐漸減小,比表面積和表面羥基數量逐漸增加。負載活性金屬Ni Mo后,由于MSI增強,MoS2片晶長度和堆疊層數逐漸降低,TypeⅠ類相占比增加,不利于渣油加氫降殘炭反應。曾雙親等[36]進一步將水熱處理時間延長至48 h,發現在90℃下水熱處理時間超過24 h時,水合作用的結合水會轉化為擬薄水鋁石晶體的結構水,并且Al2O3載體表面羥基的數量不會再隨水熱處理時間增加而變化。Liu Guangci等[37]研究了水熱處理溫度對γ-Al2O3再水合為擬薄水鋁石過程的影響。由XRD表征結果可知,隨著水熱處理溫度的升高,擬薄水鋁石的衍射峰強度增加,說明高溫水熱處理有利于γ-Al2O3再水合為擬薄水鋁石。此外,水熱處理溫度高于80℃時,Al2O3才能完全再水合為擬薄水鋁石。為了進一步明確水熱處理溫度對載體的影響,Zhang Cen等[38]將擬薄水鋁石粉在550℃下焙燒,得到的γ-Al2O3經過不同溫度水熱處理后,統一在550℃焙燒,制備出不同溫度水熱處理后的γ-Al2O3載體。研究結果表明,隨著工業γ-Al2O3的水熱處理溫度從120℃升至180℃,Al2O3晶粒逐漸增大,比表面積逐漸減小,且MSI逐漸降低,硫化后更易生成平均尺寸更大、平均堆疊層數更多的MoS2片晶,具有更高的邊角比。以噻吩(S質量分數500μg/g)、1-己烯(質量分數20%)和正庚烷組成的混合物為原料反應時,其加氫脫硫選擇性因子從1.21提高到2.51,表現出更高的加氫脫硫選擇性。
2.2 硫化條件
催化劑硫化是將催化劑活性組分從氧化態轉化為硫化態并形成活性相結構的關鍵步驟。硫化條件,包括硫化劑的類型(氣體或液體)、硫化壓力、溫度和時間等都會導致活性相形貌結構的變化。Haandel等[24]探究了硫化壓力為2.0 MPa時,分別以10%(φ)H2S/90%(φ)H2和二甲基二硫醚(DMDS)作為硫化劑對 Co Mo-CA/Al2O3催化劑活性相形貌結構的影響。在相同的硫化溫度下,經DMDS液相硫化后的催化劑,63%的MoS2片晶以單層形式存在;而經10%(φ)H2S/90%(φ)H2氣相硫化后的催化劑只有15%的MoS2片晶以單層形式存在。這是由于在兩種硫化方式下,活性金屬的起始硫化溫度不同,氣相H2S在室溫下就可以使活性金屬逐步硫化,而DMDS需在150~250℃下分解出H2S后才能使活性金屬快速硫化,分解溫度限制了MoS2片晶的生長。
Chen Jianjun等[39]在10%(φ)H2S/90%(φ)H2、400℃條件下,考察了不同硫化壓力(0.1 MPa和4.0 MPa)對 Mo/Al2O3催化劑硫化效果的影響。結果表明,在4.0 MPa下硫化生成的 MoS2片晶尺寸大于在0.1 MPa下硫化生成的MoS2片晶,能暴露出更多有助于噻吩HDS反應的S-edge位點。而對于Co Mo/Al2O3催化劑體系[40],相同硫化氛圍下,改變硫化壓力基本不影響活性片晶的尺寸,但是經過4.0 MPa高壓硫化后的催化劑易形成更多的TypeⅡ類活性相。Kooyman等[41]將1.0 MPa、400℃條件下硫化后的 Co Mo/Al2O3催化劑于0.1 MPa、600℃條件下繼續補充硫化后發現,催化劑的噻吩 HDS活性下降。從TEM中觀察到的MoS2片晶從彎曲變為筆直,但是彎曲的MoS2片晶反而更容易暴露出活性位點。
此外,倪雪華等[42]發現:在低溫(250 ℃)硫化時,NiW/Al2O3催化劑的TEM照片中存在納米尺寸的斑點狀顆粒;隨著硫化溫度升高,斑點狀顆粒消失,WS2片晶增多,且 WS2片晶的平均尺寸和平均堆疊層數顯著增加,形成更多高活性的TypeⅡ類活性相。此前,由于TEM表征手段的局限,無法對這些斑點狀顆粒進行定性,但結合何文會等[12]通過Cs-STEM表征對活性相形貌結構的新認識,推測這些斑點狀顆粒可能是單原子、金屬簇。Besenbacher等[11]研究發現,在100%(φ)H2S硫化氛圍下,隨著硫化溫度從550℃升高至700℃,負載在工業 Al2O3載體上的 Mo/Al2O3和Co Mo/Al2O3催化劑的活性相尺寸均逐漸增加。通過HR HAADF-STEM觀察到低溫硫化時存在的小于1 nm的單原子、金屬簇,經過高溫硫化后基本消失。此外,Mo/Al2O3催化劑的活性相形貌隨著硫化溫度升高由不規則形狀轉變為三角形或圓角三角形;而Co Mo/Al2O3催化劑的活性相形貌隨著硫化溫度升高由規則六邊形逐漸轉變為不規則多邊形,Co的助劑化作用導致硫化溫度對活性相形貌的影響出現差異。
基于上述研究可以看出,對于負載型加氫催化劑而言,通過調變載體的表面結構性質,可以改變MSI;減弱MSI有利于金屬物種的硫化以及活性相的生長,生成的活性片晶的尺寸更大、堆疊層數更多。另外,不同硫化條件對不同方法制備的催化劑的影響也不盡相同,需要根據目標活性相形貌結構的具體要求選擇合適的調變手段,以達到定向設計催化劑活性相形貌結構的目的。
3 活性相結構變化對催化性能的影響
活性相形貌結構的不同會帶來活性中心種類和數量的差異,而不同加氫反應對活性中心的種類和數量需求有所不同[4]。過去基于TEM只能觀察到條紋堆垛狀形貌結構的活性相片晶[43],對催化劑活性相結構與催化性能之間構效關系的研究側重于活性相片晶尺寸以及堆疊層數對催化劑性能的影響。隨著表征技術的進步,使用Cs-STEM可以區分單原子、金屬簇等更精細的活性相形貌結構,豐富了活性相形貌結構與催化劑活性之間的關系。但目前關于這些單原子、金屬簇形貌的活性相結構對催化劑活性影響的報道還較少。Alphazan等[44]結合DFT計算發現,通過HR HAADF-STEM觀察到的以無定形硅鋁材料(ASA)為載體的催化劑 WS2/ASA的活性相形貌會影響活性相上W-edge和S-edge的分布狀態。其中,六邊形形貌的催化劑活性相上同時存在著分別利于H2活化和甲苯活化的兩類活性位點,能最大限度地減少兩個反應對相同位點的競爭,使催化劑的甲苯加氫飽和性能顯著提高。Ryaboshapka等[45]通過在有S摻雜的活性炭載體(SC)上浸漬硫代鉬酸鹽制備出了活性相形貌為單原子、金屬簇的催化劑,以噻吩為模型化合物評價了催化劑的HDS活性,結果發現這些催化劑上每個Mo原子的HDS TOF值都比相同制備條件下、以Al2O3為載體的催化劑高2~3倍。這也表明了采用特定方法制備的單原子、金屬簇在反應中也能表現出較好的催化性能。
活性相結構的穩定性對催化劑性能也具有重要的影響。Zavala-Sanchez等[27]將具有大量單原子、金屬簇活性相的Co Mo/Al2O3催化劑在350℃下進行噻吩HDS反應(7.9%噻吩、90%H2以及2.1%H2S混合氣體)。反應72 h后觀察卸劑時發現,活性相形貌結構沒有發生明顯變化,說明在此反應條件下,單原子、金屬簇形貌的活性相結構具有一定的穩定性。但是,在實際情況中經過長時間工業運轉后的柴油加氫催化劑的活性相會聚集長大,甚至出現助劑金屬剝離的情況,從而破壞活性相結構,導致催化劑活性下降。Chen Wenbin等[30]發現,經高硫原料A和高氮、高多環芳烴原料B交替反應216 h后的卸出劑的活性相會聚集長大,平均尺寸從3.3 nm增至4.2 nm。相比于新鮮硫化劑,卸出劑的硫化度基本沒有變化,但是其Co-Mo-S比例較新鮮硫化劑顯著降低。He Wenhui等[13]通過Cs-STEM 觀察到經過含有2.0% (w)二甲基二硫化物(DMDS)的煤油硫化后的新鮮工業Ni Mo催化劑Cat-A中,存在著單原子、金屬簇。將Cat-A在340℃、6.40 MPa條件下,以直餾柴油(SRGO)為原料,經過48 h反應后得到催化劑Cat-B,其活性相形貌結構相比于Cat-A沒有發生明顯變化。繼續將Cat-B在370℃、3.20 MPa條件下,以50%SRGO和50%催化裂化輕循環油(LCO)為原料,反應144 h后得到催化劑Cat-C,發現其單原子、金屬簇減少,而MoS2片晶尺寸增加,出現更多的單層MoS2。該現象可能是由于反應過程中,單原子、金屬簇不斷生長變化產生的。另外,由XPS的Ni 2p譜圖發現,Cat-B和Cat-C中出現了明顯的NixSy的峰,說明Ni金屬剝離,助劑效應減弱,從而導致催化劑HDS和HDN活性有所下降。此前,Eijsbouts等[46]通過高分辨透射電鏡(HRTEM)發現,失活的NiMo/Al2O3催化劑中出現了Ni3S2,表明助劑Ni從Ni-Mo-S活性相結構中剝離,從而導致催化劑活性降低。此外,積炭也是催化劑失活的主要原因,炭的沉積不僅會堵塞催化劑孔道,還會覆蓋活性金屬,導致反應物分子與活性中心的可接近性下降。通過再生的方式能除去部分積炭并恢復催化劑的活性,但Kohli等[47]對 Co Mo/γ-Al2O3新鮮劑、脫蠟油加氫處理產生的廢劑和再生劑進行活性比較時發現,再生劑表現出低HDS活性。這可能是由于經高溫再生過程再硫化后,再生劑活性相結構由高活性的TypeⅡ類相轉變為低活性的TypeⅠ類相,導致邊緣活性位數量減少,催化劑的HDS活性降低。
綜上所述,通過Cs-STEM觀察到的催化劑的單原子、金屬簇形貌結構的活性相對催化劑活性的貢獻是毋庸置疑的。首先,單原子、金屬簇的平均尺寸小于片晶,能暴露出更多的活性中心,提高金屬利用率,增加催化劑活性。其次,單原子、金屬簇的形貌不規則,可在一定程度上改變M-edge和S-edge的相對比例關系,進而可以調變催化劑的活性和選擇性。此外,炭的沉積、活性相聚集長大以及助劑剝離都會破壞活性相的結構,從而導致柴油加氫催化劑失活。
4 總結和展望
目前基于已被廣泛接受的Co(Ni)-Mo(W)-S活性相模型,借助TEM觀測到的片晶形貌結構,已經開展了較系統的加氫催化劑構效關系研究,研究結果表明加氫催化劑的活性相結構是決定催化劑活性、選擇性和穩定性的關鍵。隨著表征技術的發展,通過Cs-STEM等技術手段在工業加氫催化劑上觀察到了單原子和金屬簇等更精細的活性相形貌結構,而基于Co(Ni)-Mo(W)-S片晶形貌結構建立的TypeⅠ類相和TypeⅡ類相結構模型均未涉及此類單原子和金屬簇信息。因此,未來應在Cs-STEM等觀測基礎上,借助EXAFS、IR-CO探針分子吸附以及密度泛函理論(DFT)計算等表征手段,重新審視之前關于活性相結構的研究工作。
另外,鑒于目前以Al2O3為載體制備的工業加氫催化劑上觀察到的單原子、金屬簇和片晶等不同形貌結構混雜且共存于載體表面,無法明確不同形貌結構活性相對催化劑性能的貢獻。未來首先需要通過設計制備出活性相的形貌結構分別為單原子、金屬簇、片晶的催化劑,然后再系統地研究單原子、金屬簇、片晶等典型形貌結構之間的相互關系以及其對催化劑性能的影響,從而進一步明確理想的活性相結構特點,豐富發展現有加氫催化劑活性相結構模型,為新型加氫催化劑的高效研發提供必要的科學依據和理論指導。
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
現代企業(2015年9期)2015-02-28 18:56:50
