不同篩選方法的低密度SNP集合填充準確性比較
2023-05-15 08:52:02林雨濃王澤昭陳燕朱波高雪張路培高會江徐凌洋蔡文濤李英豪李俊雅高樹新
中國農業科學 2023年8期
林雨濃,王澤昭,陳燕,朱波,高雪,張路培,高會江, 徐凌洋,蔡文濤,李英豪,李俊雅,高樹新
不同篩選方法的低密度SNP集合填充準確性比較

1內蒙古民族大學動物科技學院,內蒙古通遼 028042;2中國農業科學院北京畜牧獸醫研究所,北京 100193;3通遼京緣種牛繁育有限責任公司,內蒙古通遼 028006
【目的】嘗試通過在華西牛參考群高密度標記芯片位點中,使用兩種標記篩選方法挑選具有代表性的且密度梯度不同的SNP位點集合,后利用基因組填充策略在相同填充參數下將低密度芯片數據填充至高密度繼而進行后續基因組研究,從而達到降低華西牛基因型分型成本的目的。研究分別比較了不同標記集合填充準確性和填充一致性的差異,闡述了標記篩選方法、標記密度、最小等位基因頻率和參考群體數量等4個因素對填充結果的影響,為華西牛低密度SNP填充芯片設計提供參考。【方法】將質控后剩余的1 233頭華西牛群體隨機分為參考群(986頭)和驗證群(247頭)。使用等間距法(equidistance,EQ)和高MAF法(high MAF,HM)兩種標記篩選方法分別從華西牛參考群體的Illumina Bovine HD 芯片位點集合中篩選出16種不同密度的SNP集合,共生成32種不同SNP梯度密度集合。隨后在驗證群體中利用Beagle(v5.1)軟件將各低密度集合填充至770 k密度水平,計算填充準確性和填充一致性并對填充性能影響因素進行分析。【結果】32種低密度SNP集合的標記數量在100—16 000之間,窗口最大為24 176 kb,最小151 kb。隨著標記密度升高,EQ和HM兩種篩選方法填充一致性和準確性不斷提升,但填充準確性和填充一致性增加的幅度越來越小。當標記集合密度超過12 k后均趨于平穩。SNP密度在16 k時兩種方法的填充準確性達到最高(=0.8801,=0.8696)。當標記密度低于11 k時,不同標記密度梯度下HM方法填充一致性均高于EQ方法。然而當SNP集合密度超過11 k時,EQ篩選方法較表現出填充優勢。與填充一致性結果趨勢相似,在SNP集合密度低于10 k時,HM方法仍然具有較高的填充準確性,但當SNP集合密度高于10 k時,EQ方法的填充準確性則較高,且在SNP密度集合大于12 k后,EQ填充準確性趨于穩定。同時研究發現與低MAF標記位點相比,高MAF位點的填充準確性更高。填充過程中發現,填充一致性和填充準確性隨著參考群體增大而提高。當參考群體數量在600—800時,位點填充準確性和一致性較高。【結論】在華西牛群體中,填充一致性和填充準確性隨標記密度遞增而上升,在標記密度為10 k—12 k區間,可獲得較好的填充效果。當標記密度小于10 k時優先選擇HM方法,更高密度時EQ方法較好。高MAF標記位點填充準確性更高。采用填充策略進行低密度標記填充時,參考群體數量在400頭以上時填充效果較為理想。
填充準確性;低密度SNP芯片;華西牛;連鎖不平衡;最小等位基因頻率
0 引言
【研究意義】基因組選擇(genomic selection,GS)作為畜禽育種的重要技術手段備受關注[1]。高密度分型帶來的高成本是目前限制基因組選擇技術在肉牛育種中推廣應用的關鍵因素之一。通過基因型填充技術,使用較低分型成本獲得的高密度基因型數據將會對肉牛育種及相關科研工作奠定基礎。【前人研究進展】高密度SNP芯片對解析重要經濟性狀的群體遺傳學分析、基因定位、基因組選擇有著重要的意義。2007年12月,Illumina公司推出了Infinium BovineSNP50 BeadChip(BovineSNP50)商業芯片并在奶牛育種中得到了廣泛應用[2]。但HAYES等指出在跨品種評估時只有當標記密度小于10 kb時,才可能在不同品種牛群中檢測到相同LD分型[3]。BovineSNP50 芯片由于密度較低存在較大使用局限[4]。為了解決標記密度問題,Illumina公司在2010年1月推出了首款牛高密度芯片(BovineHD Genotyping BeadChip,BovineHD)[5]。CARVALHEIRO等的研究證實,相對于50k或更低密度的芯片,高密度SNP芯片可以通過連鎖不平衡(linkage disequilibrium , LD)捕獲更多的變異信息,從而獲得準確性更高的基因組估計育種值[6]。VANRADEN等研究結果同樣證實利用高密度SNP信息在進行GWAS關聯分析時,可以得到更加準確的定位結果[7]。盡管隨著當前生物技術快速發展,高密度SNP芯片的價格在不斷降低,但大范圍使用高密度SNP芯片帶來的成本問題仍然限制了基因組育種技術的廣泛應用。2003年,LI等提出基因組填充方法,獲得了個體未被分型位點的基因型數據[8]。通過先期使用低密度SNP芯片對個體進行分型,隨后使用填充技術將低密度數據填充到高密度水平為降低分型成本提供了一種可行的思路[9]。相關研究也在豬[10]、馬[11]、雞[12]等畜種中展開。【本研究切入點】增加標記密度可以提升育種值估計的準確性。在華西牛群體內針對填充準確性的研究鮮有報道。本研究利用填充策略將低密度芯片數據填充至高密度繼而進行后續基因組研究,基于該設想,使用等間距法(equidistance, EQ)和高MAF法(high MAF , HM)兩種篩選方法構建出不同梯度密度SNP集合。比較各集合填充性能的差異,探究篩選方法、標記密度、MAF和參考群體數量對填充準確性的影響。【擬解決的關鍵問題】針對華西牛群體,比較各集合填充性能的差異,探究篩選方法、標記密度、MAF和參考群體數量對填充準確性的影響。確定低密度SNP集合篩選最優參數組合,最終目的是實現填充后效果與Illumina Bovine HD芯片分型結果一致,為華西牛低密度芯片設計提供依據。
1 材料與方法
1.1 試驗群體及SNP分型
本研究所使用的數據材料均來自中國農業科學院北京畜牧獸醫研究所牛遺傳育種創新團隊組建的華西牛參考群體。該群體組建于2007年,群體分布于內蒙古錫林郭勒盟烏拉蓋管理區。經過逐漸擴群,至2020年該群體共包含華西牛1 478頭,每頭均有87個表型測定記錄、系譜記錄及770 k高密度芯片分型數據。每年7—8月份,對基礎群母牛及犢牛進行基礎數據測定,10月份將育肥犢牛集中轉運至北京或圍場育肥場進行集中育肥,飼料配方保持一致。集中育肥期間每3個月全群測定一次體重和體尺數據。集中育肥6個月時,靜脈采血20 mL,并取2 mL血液進行DNA提取工作,使用Illumina Bovine HD BeadChip芯片進行基因分型。
1.2 數據質控
使用PLINK v1.90[13]軟件對常染色體基因型數據進行質量控制,質控順序為先位點后個體。具體質控標準如下:一是刪除基因分型檢出率小于90%,最小等位基因頻率小于0.01和哈代溫伯格平衡檢驗值小于1×10﹣6的SNP位點;二是刪除SNP缺失率大于10%的個體。質控后獲得1 233頭個體和671 164個SNP標記用于后續研究。
1.3 構建SNP集合
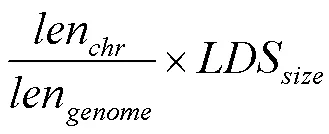
1.4 基因型填充及填充準確性評估
使用Beagle(v5.1)軟件對32個SNP集合進行填充[16]。Beagle的填充程序分兩階段進行,首先對所有待填充SNP集合進行基因定相[17],隨后進行基因型填充。所有SNP集合的定相和填充均采用Beagle的默認參數進行。
基因型填充時隨機選擇總群體20%的個體(247頭)作為驗證群體,剩余80%的個體(986頭)作為填充參考群體。在計算參考群體大小對填充準確性的影響時,驗證群體大小為總群的20%,在剩余個體中分別抽取100、200、400、600和800頭組成參考群體,計算標記密度在7、10、13和16k時的填充結果。
采用填充一致性(concordance rate, CR)和填充準確性(imputation accuracy,2)[18]衡量基因型填充效果。其中填充一致性定義為驗證群體中正確填充的位點數目與全部填充的位點數目的比值[19];填充準確性為填充位點的基因型(012編碼)和真實基因型間的相關系數平方[20]。
2 結果
2.1 不同標記密度SNP集合篩選
使用EQ和HM兩種位點篩選方法,在16組密度梯度中篩選出32個SNP集合。不同SNP集合包含的位點數量及窗口大小見表1。SNP集合中窗口最大為24 176 kb,最小為151 kb。
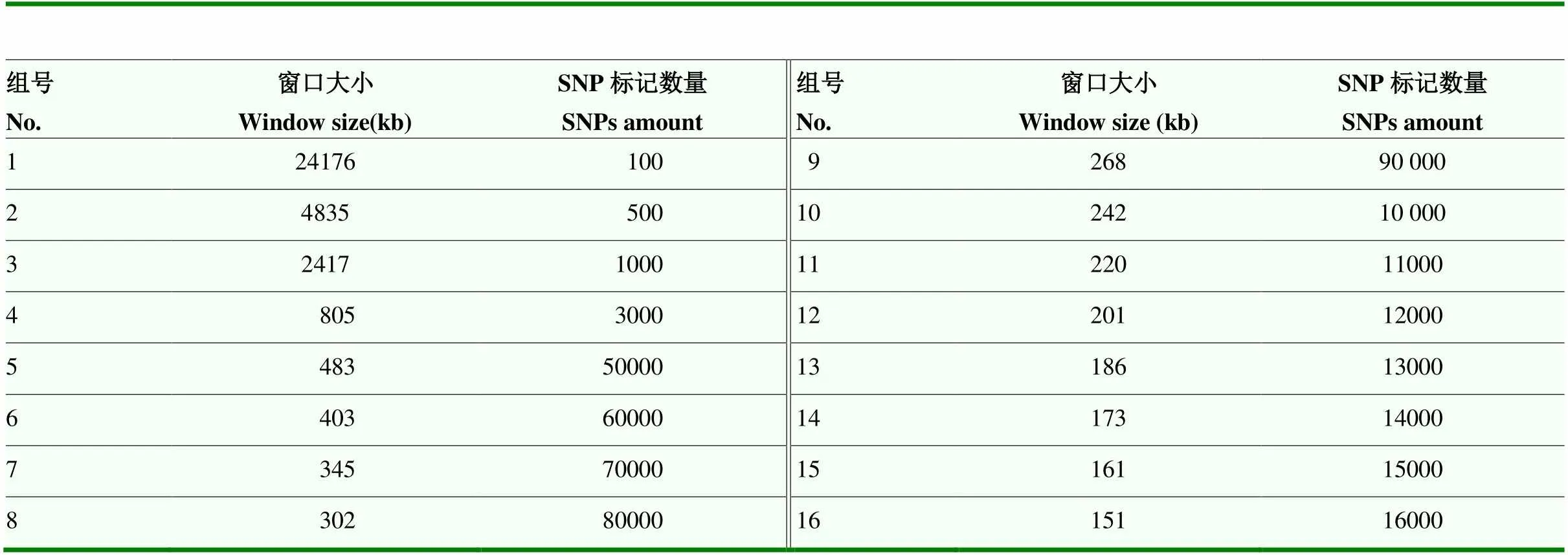
表1 不同密度集合窗口大小及SNP數量
2.2 標記密度對填充效果的影響
圖1展示了EQ和HM方法篩選出的SNP集合的填充一致性和填充準確性。如圖所示,EQ的填充一致性范圍為0.5701—0.8863,HM的填充一致性范圍為0.5743—0.8872。填充準確性的范圍與填充一致性相似,EQ方法變化范圍為0.0304—0.8801,HM方法變化范圍為0.0393—0.8696。綜合來看,隨著SNP集合密度越來越大,填充一致性和填充準確性也隨之增加,但SNP集合密度越高,填充準確性和填充一致性增加的幅度越來越小。SNP集合密度超過12 k后填充準確性和填充一致性變化趨于穩定,以EQ方法為例,從12 k到16 k增加了1.3%(填充一致性)和1.92%(填充準確性)。不同密度SNP集合的填充準確性和填充一致性結果見附表1。
2.3 標記篩選方法對填充效果的影響
如圖1所示,EQ和HM兩種方法的填充一致性和填充準確性在不同密度時存在差異。標記密度在11 k以下時HM方法具有較高的填充一致性和填充準確性,在標記密度大于11 k時EQ方法略好于HM方法。與填充一致性結果趨勢相似,在SNP集合密度低于10 k時,HM方法仍然具有較高的填充準確性,但當SNP集合密度高于10 k時,EQ方法的填充準確性則較高,且在SNP密度集合大于12 k后,EQ填充準確性趨于穩定。
2.4 最小等位基因頻率對填充效果的影響
填充準確性和填充一致性均與最小等位基因頻率有關。圖2中展示了標記密度為16 k時,EQ和HM篩選方法中填充一致性和填充準確性與標記MAF的分布。當MAF<0.1時,填充準確性較低但填充一致性較高。隨著標記自身MAF的增加,在EQ篩選方法中,平均填充一致性平均值由0.9632下降到0.8276,填充準確性平均值由0.8533上升到0.8890;在HM篩選方法中,填充一致性平均值由0.9584下降到0.8508,填充準確性平均值由0.8220上升到0.8976(圖3)。EQ和HM方法不同MAF區間填充一致性均值和填充準確性均值見附表2。
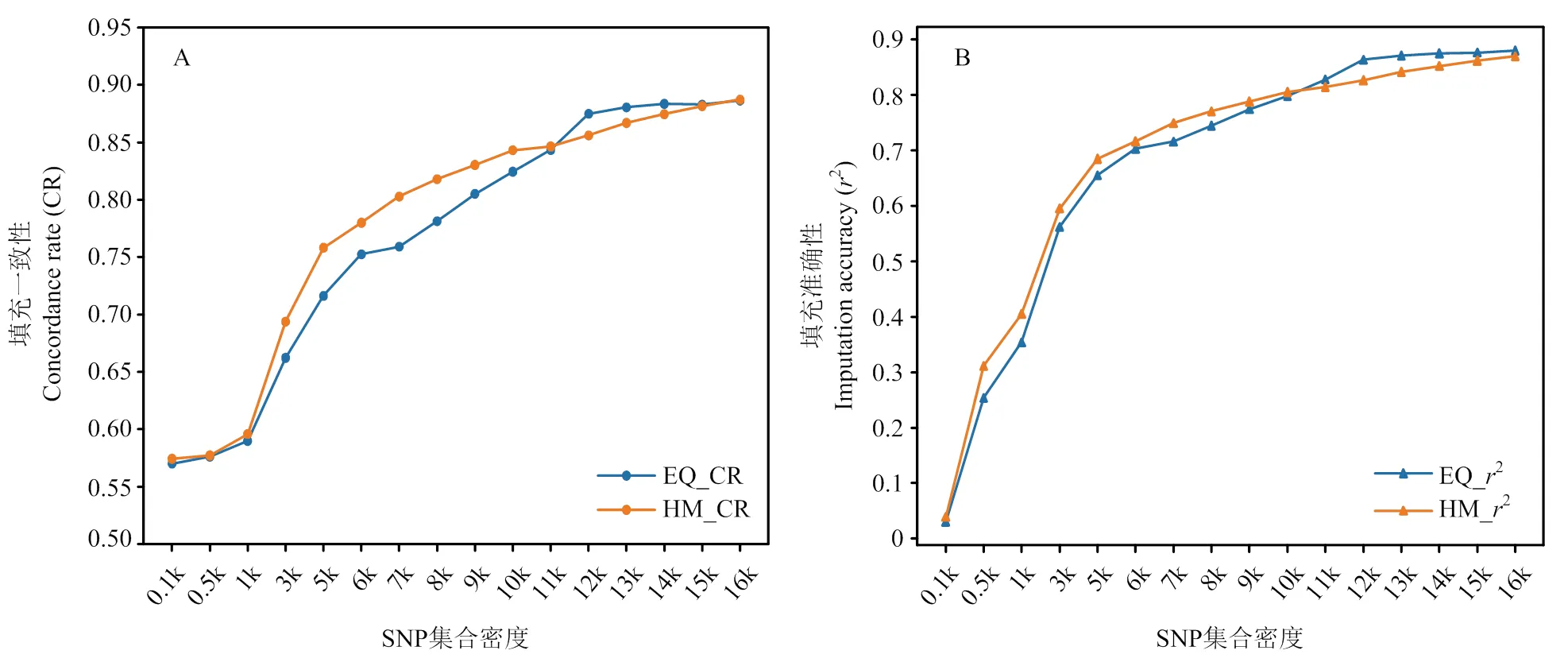
圖1 不同篩選方法的填充一致性(A)和填充準確性(B)
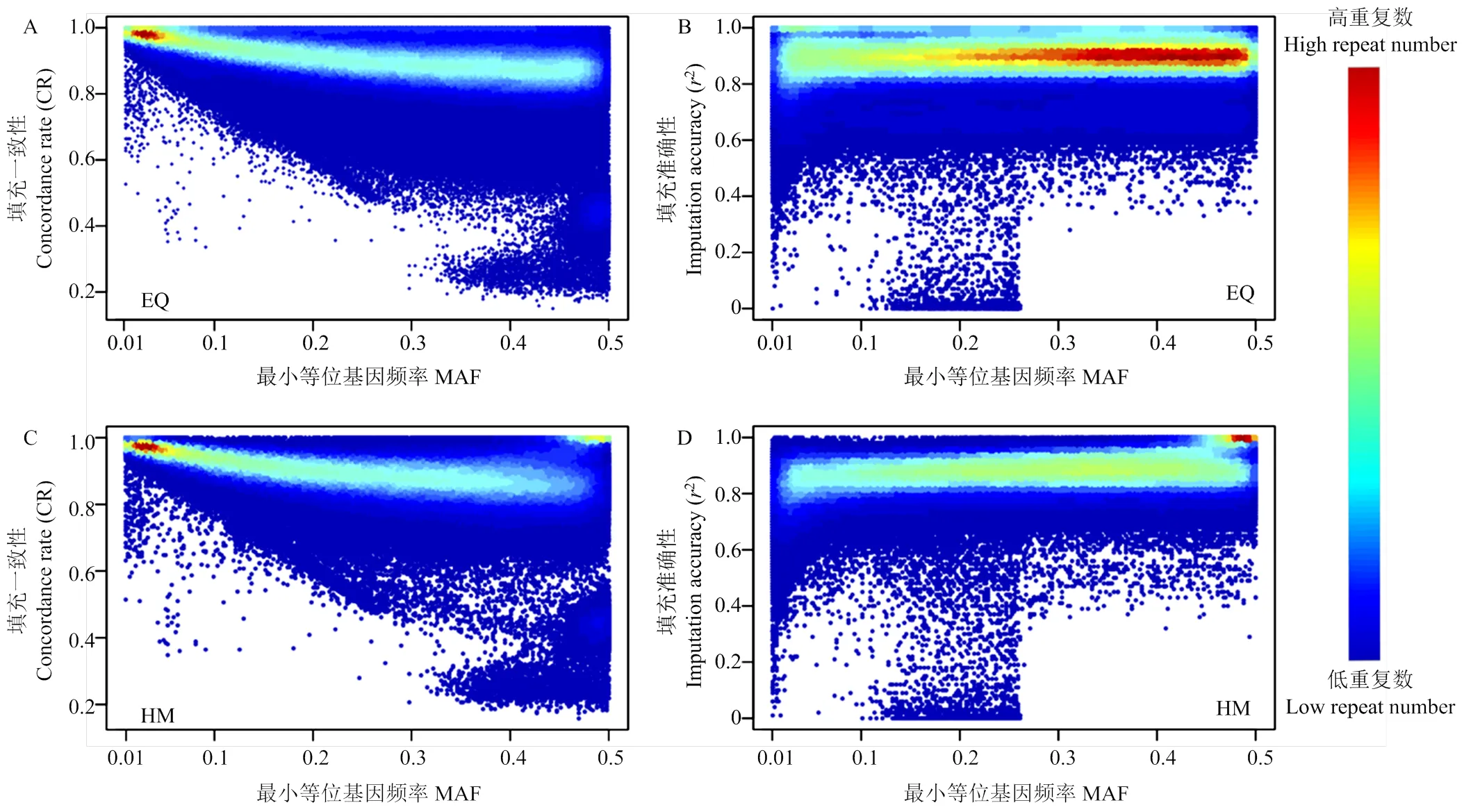
A:EQ篩選方法的填充一致性;B:EQ篩選方法的填充準確性;C:HM篩選方法的填充一致性;D:HM篩選方法的填充準確性。紅色代表相同結果的位點數量多,藍色代表相同結果的位點數量少
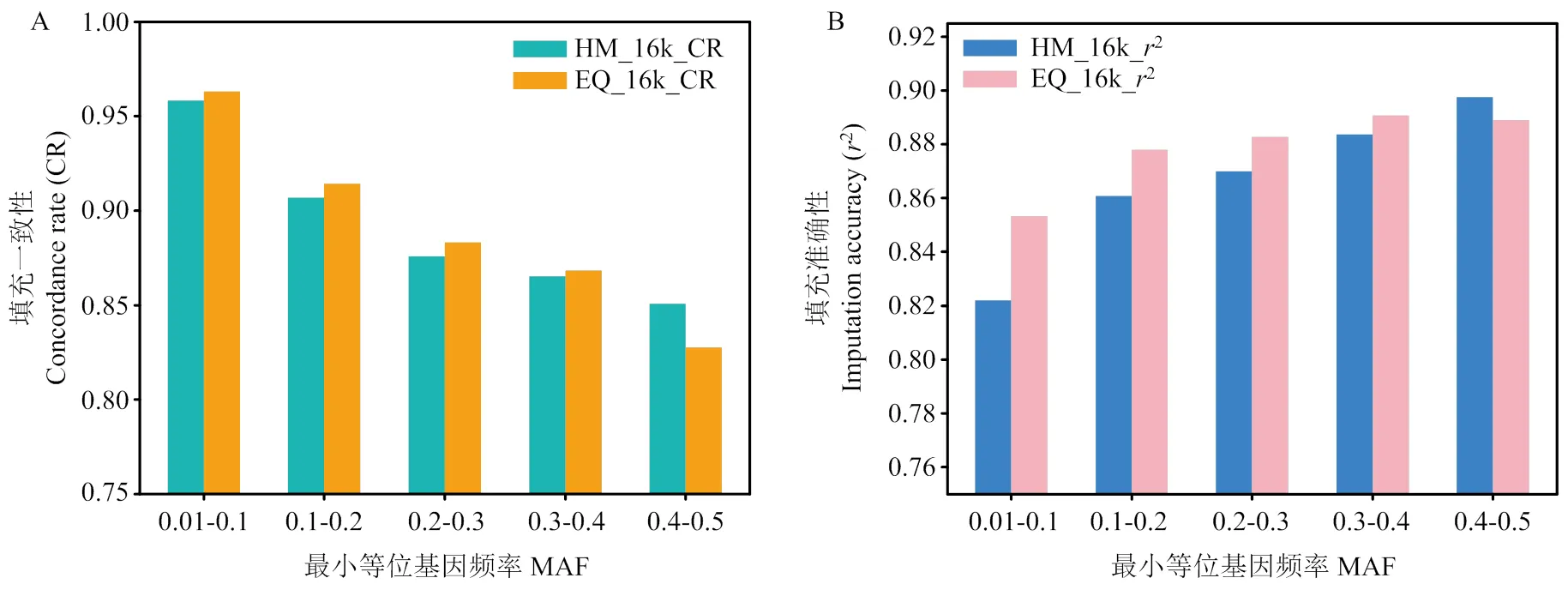
圖3 密度為16 k時兩種篩選方法的填充一致性平均值(A)和填充準確性平均值(B)與最小等位基因頻率
2.5 參考群體對填充效果的影響
分別計算了標記密度在7、10、13和16 k時,參考群體大小分別為100、200、400、600和800時的填充一致性和準確性(圖4)。結果發現,填充一致性和填充準確性隨著參考群體增大而提高。以EQ篩選方法16 k密度為例,隨著參考群體數量增加,相鄰兩個梯度之間的填充一致性增長率為:6.91%(100—200)、5.79%(200—400)、2.45%(400—600)和1.07%(600—800)。填充準確性增長率為:7.19%(100—200)、5.52%(200—400)、2.26%(400—600)和0.97%(600—800)。當參考群體數量在100—400頭時,填充一致性和準確性的增速最快。當參考群體數量超過400時,填充一致性和填充準確性提升速度開始降低。當參考群體規模為800時,不同密度SNP集合填充一致性和填充準確性達到最高,填充一致性范圍為0.7511(7 k)到0.8797(16 k)、填充準確性范圍為0.7492(7 k)到0.8750(16 k),見附表3。
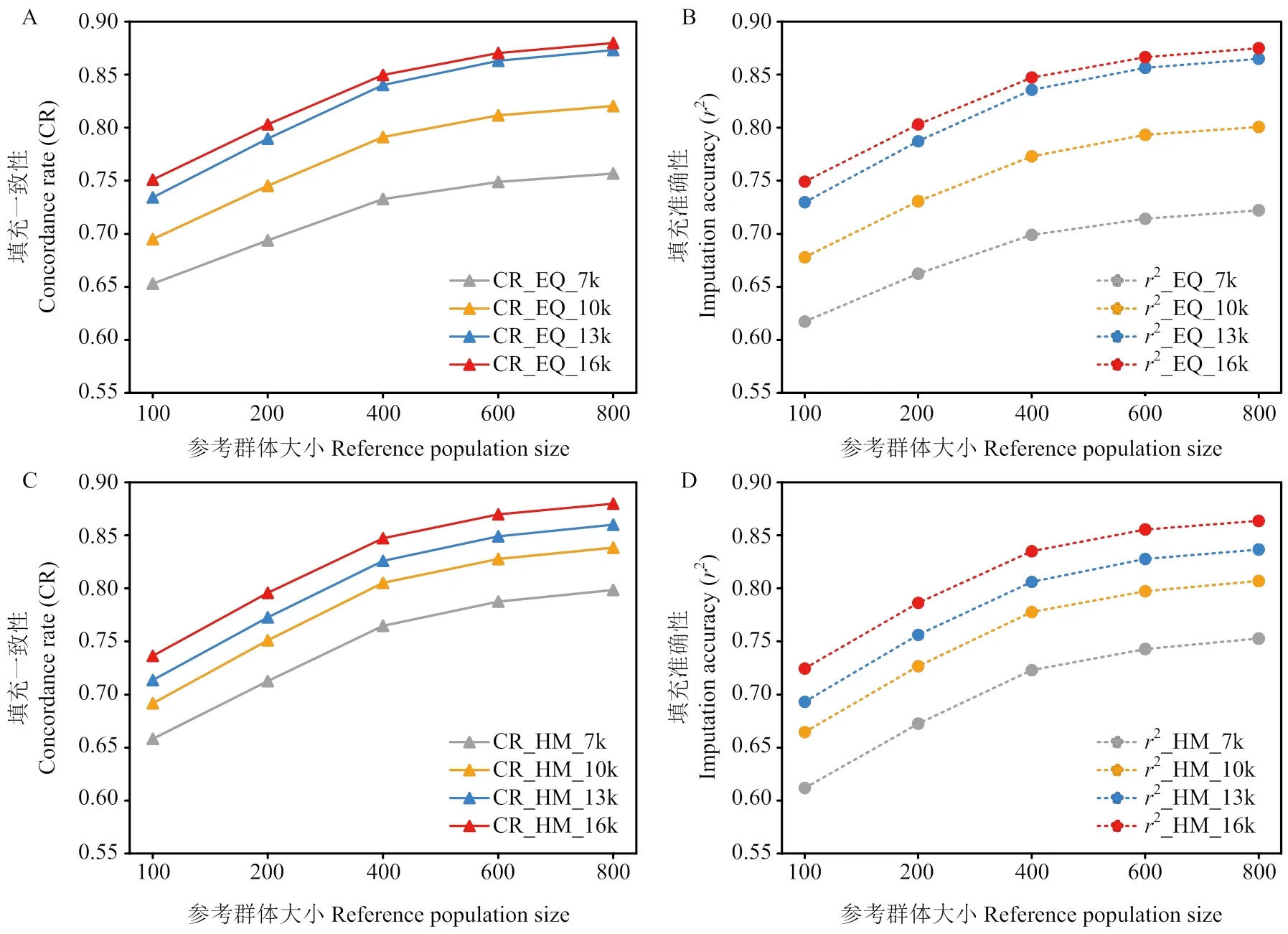
圖4 參考群體大小對填充準確性和填充一致性的影響
3 討論
3.1 標記密度對填充的影響
本研究目的是探究華西牛群體低密度SNP集合填充到770 k密度的填充效果。發現,低密度SNP集合的填充一致性和準確性均隨標記密度增加而升高。在不同密度下SNP集合的填充一致性的提升趨勢有顯著差別,呈現出慢-快-慢的增長曲線。以EQ方法填充一致性為例,在密度較低時提升緩慢,從0.1 k到1 k僅提升3.51%。密度在1 k至12 k時提升較快,增長了48.31%。密度大于12 k后提升較慢,12 k到16 k僅提升1.30%。CALUS等報道,隨著SNP芯片中標記不斷增加,對應的參考單倍型將更加容易被識別出來,容易獲得更高的填充準度[21]。因此可以推斷,SNP集合密度對于填充效果的影響,本質上是基因組上單倍型的分布情況導致的。隨著SNP標記密度增加,落在單倍型上的概率變大,連鎖出附近其他位點的能力增強,因此填充準確性和一致性變高。
在本研究中,當集合密度大于12 k時,EQ和HM篩選方法的填充準確性和一致性均趨于穩定,因此推斷在華西牛群體中最佳SNP填充密度標記在12 k左右。相關研究指出最佳SNP填充密度可能與基因組大小有關。WENG 等報道,在荷斯坦奶牛中,使用標記密度為7 k的芯片進行分型后,通過填充就可以獲得較高填充準性的高密度SNP集合[22]。WANG等在關于雞的研究中發現[23],同種群體中包含約400個均勻分布的標記,使用低密度芯片進行GEBV估計的準度相比于高密度芯片僅損失了6%。WELLMANN研究發現,在豬中使用含有3 000個位點的低密度芯片估算的GEBV,與密度為60 k的 SNP芯片估計得到的GEBV結果具有高度相關[24]。因此,在后續研究中可以繼續探索在此密度上填充到高密度水平后的估計育種值準確性。
3.2 均勻分布對于填充的影響
當標記密度低于10 k時,HM方法具有較高的填充一致性和填充準確性。推斷在標記密度低時,高MAF位點與周圍QTL之間處于連鎖不平衡狀態的概率更高。此密度下使用EQ方法構建低密度SNP集合的填充準確性和一致性均較低。這是由于標記密度過低,相鄰位點之間的連鎖情況較差,從而使Beagle軟件利用位點間的連鎖進行填充的算法不能發揮出最優作用。當標記密度高于10 k時,EQ策略優于HM策略,其原因是等間距的篩選位點可以在位點數量有限的情況下,最大限度的利用分布在整個基因組上的遺傳信息,因而相較于HM方法更具有優勢。
3.3 最小等位基因頻率對于填充的影響
本研究結果顯示,隨著標記自身MAF增加,填充一致性均值逐漸下降,填充準確性均值逐漸增加。同時觀察到,在MAF小于0.1時,填充準確性較低。關于MAF對填充準確性的影響,在不同文章存在完全相反的觀察結果,但均未深入展開討論。Didier等針對Illuminate BovineLD芯片的研究結果表明,在多數肉牛品種中SNP位點的MAF高于0.3,在該情況下觀察到高MAF位點有助于提高填充的準確性[14]。HERRY等同樣利用等間距法挑選SNP位點用于填充,同樣發現,低MAF的SNP位點填充錯誤率更高[25]。這可能是由于低MAF位點導致該位點的分型錯誤率提升,進而導致的填充錯誤率提高[26]。
但同樣有研究報道,隨著位點MAF的提升,填充準確性降低或對填充準確性沒有影響。HOZé等在Montbéliarde品種的填充研究中,根據填充準確性將SNP分為兩類。第一類是對于填充準確性大于0.9的SNP位點,沒有觀察到填充準確性與MAF存在相關關系。第二類對于填充準確性小于0.9的SNP位點,填充準確性隨著MAF的增加而降低,他們猜測這可能與SNP位點的錯誤定位(miss mapping)有關,但未展開深入討論[27]。羅漢鵬等使用Beagle 5.1和Minimac 3兩款填充軟件對荷斯坦奶牛基因組50 k芯片數據填充至150 k時,同樣發現隨著最小等位基因頻率的增加,兩款填充軟件的填充一致性都表現較差,尤其平均最小等位基因頻率在0.4—0.5時(位點約占待填充位點的29%),填充準確性相對較低[28]。上述兩種觀察結果與本研究相似。
3.4 參考群體對填充的影響
參考群體數量是影響填充效果的重要因素之一,在豬、牛、羊和雞等不同物種類似研究中均有報道[21, 29-30]。本研究分別計算了在不同標記密度梯度下,參考群體數量分別為100、200、400、600和800時的填充一致性和填充準確性。
通過計算EQ方法16 k時參考群體數量對填充一致性的影響發現,當參考群體數量在100—400頭時,填充一致性增長最快,分別提升了6.91%(100—200)和5.79%(200—400)。當參考群體的數量超過400時,填充一致性增速開始放緩,為2.45%(400—600)。當參考群體數量大于600時,填充一致性增長率僅為1.07%(600—800)。研究發現,參考群體對填充一致性和填充準確性的影響與密度因素的影響類似。當參考群體中華西牛數量不斷增加時,填充一致性和填充準確性大幅提高,隨著參考群體數量的增加,增速不斷下降。上述結果表明,參考群體數量要維持在一個必要的大小,才能獲得理想的低密度SNP芯片填充準確性。HOZé等報道,當參考群體數量小于400時,參考群體數量是影響填充效果最主要的因素[27]。隨著參考群體數量的增加,可參考的單倍型庫的規模也隨之增加,在參考單倍型庫中發現候選單倍型片段的概率隨之增加[31-32]。這與UEMOTO等[33]、VENTURA等[18]和CALUS等[21]的結果一致。
4 結論
本研究使用EQ和HM兩種標記篩選方法,評估了華西牛群體不同密度SNP集合填充到770 k的填充結果。研究發現填充一致性(CR)和填充準確性(2)受標記密度、最小等位基因頻率、參考群體大小影響。填充一致性和填充準確性隨密度遞增而上升,在10—12 k區間內較適合用于填充,密度小于10 k時HM方法較好,密度大于10 k時EQ方法較好。低MAF位點對填充準確性的影響比較大。參考群體中個體數超過400時填充效果較好。
[1] 朱波, 王延暉, 牛紅, 陳燕, 張路培, 高會江, 高雪, 李俊雅, 孫少華. 畜禽基因組選擇中貝葉斯方法及其參數優化策略. 中國農業科學, 2014, 47(22): 4495-4505. doi:10.3864/j.issn.0578-1752.2014. 22.015.
ZHU B, WANG Y H, NIU H, CHEN Y, ZHANG L P, GAO H J, GAO X, LI J Y, SUN S H. The strategy of parameter optimization of Bayesian methods for genomic selection in livestock. Scientia Agricultura Sinica, 2014, 47(22): 4495-4505. doi:10.3864/j.issn.0578- 1752.2014.22.015. (in Chinese)
[2] VANRADEN P M, VAN TASSELL C P, WIGGANS G R, SONSTEGARD T S, SCHNABEL R D, TAYLOR J F, SCHENKEL F S. Invited Review: reliability of genomic predictions for North American Holstein bulls. Journal of Dairy Science, 2009, 92(1): 16-24.
[3] DE ROOS A P W, HAYES B J, SPELMAN R J, GODDARD M E. Linkage disequilibrium and persistence of phase in Holstein-Friesian, jersey and Angus cattle. Genetics, 2008, 179(3): 1503-1512.
[4] HAYES B J, BOWMAN P J, CHAMBERLAIN A C, VERBYLA K, GODDARD M E. Accuracy of genomic breeding values in multi- breed dairy cattle populations. Genetics, Selection, Evolution, 2009, 41: 51.
[5] MATUKUMALLI L K,SCHROEDER S,DENISE S, SONSTEGARD T, LAWLEY C T, GEORGES M. Analyzing LD blocks and CNV segments in cattle: Novel genomic features identified using the BovineHD BeadChip. 2011. www.scienceopen.com/document?vid= 0fb91f10-7679-4ec4- b5a9- ca39bd541f2e.
[6] CARVALHEIRO R, BOISON S A, NEVES H H R, SARGOLZAEI M, SCHENKEL F S, UTSUNOMIYA Y T, O'BRIEN A M P, S?LKNER J, MCEWAN J C, VAN TASSELL C P, SONSTEGARD T S, GARCIA J F. Accuracy of genotype imputation in nelore cattle. Genetics, Selection, Evolution, 2014, 46: 69.
[7] VANRADEN P M, NULL D J, SARGOLZAEI M, WIGGANS G R, TOOKER M E, COLE J B, SONSTEGARD T S, CONNOR E E, WINTERS M, VAN KAAM J B C H M, VALENTINI A, VAN DOORMAAL B J, FAUST M A, DOAK G A. Genomic imputation and evaluation using high-density Holstein genotypes. Journal of Dairy Science, 2013, 96(1): 668-678.
[8] LI N, STEPHENS M. Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics, 2003, 165(4): 2213-2233.
[9] DRUET T, SCHROOTEN C, DE ROOS A P W. Imputation of genotypes from different single nucleotide polymorphism panels in dairy cattle. Journal of Dairy Science, 2010, 93(11): 5443-5454.
[10] GROSSI D A, BRITO L F, JAFARIKIA M, SCHENKEL F S, FENG Z. Genotype imputation from various low-density SNP panels and its impact on accuracy of genomic breeding values in pigs. Animal, 2018, 12(11): 2235-2245.
[11] CORBIN L J, KRANIS A, BLOTT S C, SWINBURNE J E, VAUDIN M, BISHOP S C, WOOLLIAMS J A. The utility of low-density genotyping for imputation in the Thoroughbred horse. Genetics, Selection, Evolution: GSE, 2014, 46(1): 9.
[12] YE S P, YUAN X L, LIN X R, GAO N, LUO Y Y, CHEN Z M, LI J Q, ZHANG X Q, ZHANG Z. Imputation from SNP chip to sequence: a case study in a Chinese indigenous chicken population. Journal of Animal Science and Biotechnology, 2018, 9: 30.
[13] CHANG C C, CHOW C C, TELLIER L C, VATTIKUTI S, PURCELL S M, LEE J J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience, 2015, 4(1): s13742-15.
[14] BOICHARD D, CHUNG H, DASSONNEVILLE R, DAVID X, EGGEN A, FRITZ S, GIETZEN K J, HAYES B J, LAWLEY C T, SONSTEGARD T S, VAN TASSELL C P, VANRADEN P M, VIAUD-MARTINEZ K A, WIGGANS G R, CONSORTIUM B L. Design of a bovine low-density SNP array optimized for imputation. PLoS One, 2012, 7(3): e34130.
[15] BOLORMAA S, GORE K, VAN DER WERF J H J, HAYES B J, DAETWYLER H D. Design of a low-density SNP chip for the main Australian sheep breeds and its effect on imputation and genomic prediction accuracy. Animal Genetics, 2015, 46(5): 544-556.
[16] BROWNING B L, ZHOU Y, BROWNING S R. A one-penny imputed genome from next-generation reference panels. The American Journal of Human Genetics, 2018, 103(3): 338-348.
[17] MARCHINI J, HOWIE B. Genotype imputation for genome-wide association studies. Nature Reviews Genetics, 2010, 11(7): 499-511.
[18] VENTURA R V, MILLER S P, DODDS K G, AUVRAY B, LEE M, BIXLEY M, CLARKE S M, MCEWAN J C. Assessing accuracy of imputation using different SNP panel densities in a multi-breed sheep population. Genetics, Selection, Evolution: GSE, 2016, 48(1): 71.
[19] O’BRIEN A C, JUDGE M M, FAIR S, BERRY D P. High imputation accuracy from informative low-to-medium density single nucleotide polymorphism genotypes is achievable in sheep1. Journal of Animal Science, 2019, 97(4): 1550-1567.
[20] BROWNING S R, BROWNING B L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. The American Journal of Human Genetics, 2007, 81(5): 1084-1097.
[21] CALUS M P L, BOUWMAN A C, HICKEY J M, VEERKAMP R F, MULDER H A. Evaluation of measures of correctness of genotype imputation in the context of genomic prediction: a review of livestock applications. Animal, 2014, 8(11): 1743-1753.
[22] WENG Z, ZHANG Z, ZHANG Q, FU W, HE S, DING X. Comparison of different imputation methods from low- to high-density panels using Chinese Holstein cattle. Animal, 2013, 7(5): 729-735.
[23] WANG C, HABIER D, PEIRIS B L, WOLC A, KRANIS A, WATSON K A, AVENDANO S, GARRICK D J, FERNANDO R L, LAMONT S J, DEKKERS J C M. Accuracy of genomic prediction using an evenly spaced, low-density single nucleotide polymorphism panel in broiler chickens. Poultry Science, 2013, 92(7): 1712-1723.
[24] WELLMANN R, PREU? S, THOLEN E, HEINKEL J, WIMMERS K, BENNEWITZ J. Genomic selection using low density marker panels with application to a sire line in pigs. Genetics, Selection, Evolution: GSE, 2013, 45(1): 28.
[25] HERRY F, HéRAULT F, PICARD DRUET D, VARENNE A, BURLOT T, LE ROY P, ALLAIS S. Design of low density SNP chips for genotype imputation in layer chicken. BMC Genetics, 2018, 19(1): 108.
[26] YUAN M, FANG H Y, ZHANG H. Correcting for differential genotyping error in genetic association analysis. Journal of Human Genetics, 2013, 58(10): 657-666.
[27] HOZé C, FOUILLOUX M N, VENOT E, GUILLAUME F, DASSONNEVILLE R, FRITZ S, DUCROCQ V, PHOCAS F, BOICHARD D, CROISEAU P. High-density marker imputation accuracy in sixteen French cattle breeds. Genetics, Selection, Evolution, 2013, 45: 33.
[28] 羅漢鵬, 竇金煥, 安濤, 陳少侃, 王雅春. 基于荷斯坦牛群體基因組數據填充軟件的準確性比較(Minimac 3與Beagle 5.1). 中國畜牧獸醫, 2021, 48(5): 1664-1671.
LUO H P, DOU J H, AN T, CHEN S K, WANG Y C. Comparison of software (minimac 3 and beagle 5.1) for genomic imputation using Holstein cow population. China Animal Husbandry & Veterinary Medicine, 2021, 48(5): 1664-1671. (in Chinese)
[29] BOLORMAA S, CHAMBERLAIN A J, KHANSEFID M, STOTHARD P, SWAN A A, MASON B, PROWSE-WILKINS C P, DUIJVESTEIJN N, MOGHADDAR N, VAN DER WERF J H, DAETWYLER H D, MACLEOD I M. Accuracy of imputation to whole-genome sequence in sheep. Genetics, Selection, Evolution, 2019, 51(1): 1.
[30] HAYES B J, BOWMAN P J, DAETWYLER H D, KIJAS J W, VAN DER WERF J H J. Accuracy of genotype imputation in sheep breeds. Animal Genetics, 2012, 43(1): 72-80.
[31] VENTURA R V, LU D, SCHENKEL F S, WANG Z, LI C, MILLER S P. Impact of reference population on accuracy of imputation from 6K to 50K single nucleotide polymorphism chips in purebred and crossbreed beef cattle1. Journal of Animal Science, 2014, 92(4): 1433-1444.
[32] HEIDARITABAR M, CALUS M P L, VEREIJKEN A, GROENEN M A M, BASTIAANSEN J W M. Accuracy of imputation using the most common sires as reference population in layer chickens. BMC Genetics, 2015, 16: 101.
[33] UEMOTO Y, SASAKI S, SUGIMOTO Y, WATANABE T. Accuracy of high-density genotype imputation in Japanese Black cattle. Animal Genetics, 2015, 46(4): 388-394.
Comparison of Imputation Accuracy for Different Low-Density SNP Selection Strategies
1College of Animal Science and Technology, Inner Mongolia University for the Nationalities, Tongliao 028042, Inner Magnolia;2Institute of Animal Sciences, Chinese Academy of Agriculture Sciences, Beijing 100193;3Tongliao Jingyuan Breeding Cattle Breeding LLC, Tongliao 028006, Inner Magnolia
【Objective】To facilitate the low-cost genomic selection in Huaxi Cattle, the present study represented the first attempt to designed a new low-density Genotype chip to support imputation to higher density genotypes. The representative SNP markers with different density gradients were selected from high-density SNP chips in the Huaxi cattle reference population by using two SNP selection methods. And then, these marker sets were imputed to high-density sets with the same imputation parameters for subsequent genomic studies. Meanwhile, the current study compared the differences in imputation accuracy and concordance among SNP panels and illustrated the effects of four factors on imputation results, including marker screening method, marker density, minor allele frequency, and the number of reference population. This study could provide insights about the methods to select the low-density SNP markers for imputation in the current population and the representative SNPs, and aid in designing low-density SNP chip for Huaxi cattle.【Method】Totally 1,233 Huaxi cattle after genotypes filtered was randomly divided into reference (986) and validation (247) populations. , Based on Equidistance (EQ) and high MAF (HM), two SNP selection strategies were used to make 16 SNP sets with different densities from the Illumina Bovine HD chip in the reference population, respectively. Each of the 32 low-density set was then imputed to the 770K density level in the validation population by using Beagle (v5.1), while the imputation accuracy and concordance were calculated as the mean correlation between true and imputed genotypes. Finally, a comprehensive set of factors that influence the imputation performance were analyzed.【Result】The number of markers in the 32 low-density SNP sets ranged from 100 to 16 000, with a maximum window of 24 176 kb and a minimum window of 151 kb. The imputation accuracy and concordance of both EQ and HM methods went up with increasing marker densities. The imputation accuracy of both methods was the highest at 16k SNP density (=0.8801,=0.8696). When the marker density was below 11k, the imputation concordance of HM was higher than EQ for all marker density gradients. However, when the SNP density exceeded 11 k, EQ showed an imputation accuracy advantage over HM. Similar to the imputation concordance results, the HM method still had higher imputation accuracy when the SNP density was lower than 10 k, but the EQ method had higher imputation accuracy when the SNP pool density was higher than 10 k, and the EQ imputation accuracy tended to be stable after the SNP density was greater than 12 k. It was also found that the imputation accuracy of high MAF locus was higher. During the imputation process, it was found that the imputation accuracy and concordance increased with the increase of the reference panel. The imputation accuracy and concordance of loci were higher when the population of the reference panel was 600-800. 【Conclusion】In the Huaxi cattle population, the imputation accuracy and concordance increased with increasing marker density, and a better imputation effect could be obtained in the marker density of 10 k-12 k interval. The HM method was preferred when the marker density was less than 10 k, and the EQ method was better at high marker density. High MAF loci were more accurate for imputation. When the imputation strategy for low-density marker imputation was used, the number of reference panel should be at least 400 heads for better imputation effect.
imputation accuracy; low density SNP array; Chinese Simmental cattle; linkage disequilibrium; MAF
10.3864/j.issn.0578-1752.2023.08.013
2021-12-13;
2022-03-24
內蒙古自治區第五批“草原英才”工程產業創新創業人才團隊專項、內蒙古自然科學基金面上項目(2019MS03077)、內蒙古自治區科技計劃項目(KJXM2020002-05)、青年科學基金(32102505)
林雨濃,E-mail:Lin_Yunong@outlook.com。通信作者高樹新,E-mail:shuxingao@126.com。通信作者李俊雅,E-mail:lijunya@caas.cn。
(責任編輯 林鑒非)
猜你喜歡
公民與法治(2022年5期)2022-07-29 00:47:28
教學考試(高考物理)(2021年5期)2021-11-08 10:31:22
歷史教學問題(2021年4期)2021-11-05 07:02:34
中醫眼耳鼻喉雜志(2021年1期)2021-07-22 07:38:14
兒童故事畫報(2019年5期)2019-05-26 14:26:14
中國公共安全(2017年11期)2017-02-06 05:28:08
Coco薇(2016年2期)2016-03-22 02:42:52
燕山大學學報(2015年4期)2015-12-25 02:19:49
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
