機器學習勢在含能材料分子模擬中的研究進展
2023-06-13 10:17:42常曉雅文明杰王永錦初慶釗陳東平
火炸藥學報 2023年5期
常曉雅,文明杰,張 迪,王永錦,初慶釗,朱 通,陳東平
(1.北京理工大學 爆炸科學與技術重點實驗室,北京 100081;2.上海市分子治療與新藥開發工程研究中心, 華東師范大學 化學與分子工程學院,上海 200062)
引 言
含能材料通常指在受到外界刺激時能快速分解并釋放巨大能量的化合物或混合物[1-2],被廣泛應用于軍事燃燒及爆破、火箭導彈發射、推進劑等領域。在推進劑領域,含能材料以燃燒的形式釋放能量,實現了從化學能向動能的有效轉化,成為先進導彈武器發動機的動力來源,是推動武器裝備系統發展的支撐和制約技術之一[3-4];在火炸藥領域,含能材料在極短的時間內釋放大量的熱能和氣體,以爆轟形式作功形成沖擊波,是武器裝備完成發射和高效毀傷的能源材料。作為現代武器系統的主要能量來源,含能材料燃燒與爆炸性能直接影響武器裝備的打擊范圍和毀傷能力,是先進武器裝備優化設計的關鍵發展方向。
含能材料釋放的能量主要來源于原子核外層電子在反應過程中轉移所釋放的能量[5],其宏觀特性本質上是由其微觀分子結構所決定的。例如,含能材料爆轟反應放熱量取決于產物分子的生成熱;含能材料的感度與分子結構、分子間相互作用以及反應活化能等性質有關;各組分之間的相容性與分子間的相互作用有關[6-7]。因此,在微觀結構層面上研究含能分子結構燃燒特性和力學穩定性對于新型含能材料的設計至關重要。分子模擬作為原子尺度的研究方法,可以用來預測含能材料的力學行為和化學反應過程,揭示分子結構與宏觀燃燒性能之間的關系,從而為含能材料的理論設計提供科學依據。在分子模擬計算中,描述微觀體系中不同分子構型與能量的映射關系被稱為勢能面。分子模擬的實質是在被模擬材料分子體系的勢能面上進行動力學研究,例如搜尋分子的最低能量構形或者計算化學反應速率。其中構建勢能面的計算方法主要包括第一性原理計算和分子動力學。第一性原理計算是通過求解薛定諤方程來描述粒子的電子結構,在不需要任何先驗參數情況下,準確計算分子、原子的理化性質,計算精度較高。在空間尺度較小且精度要求高的場景,比如少量分子體系的化學反應路徑研究、動力學參數計算等,第一性原理計算表現出獨特的優勢。然而高精度的計算水平伴隨著高昂的計算成本,時間復雜度往往是O(N3)甚至更高,現有的計算能力無法承載超過1 000個原子的分子體系。
分子動力學不考慮電子運動的自由度,電子效應體現在原子間相互作用的勢函數中,原子的運動遵循牛頓第二定律,并據此實現目標體系在給定初始條件下隨時間的動態演化。分子模擬的精度取決于勢函數對真實勢能面重建的精度。勢函數的數學形式和分子各部分對能量貢獻的可拓展性等均對勢能面重建精度有影響[8]。非反應力場假設的函數形式簡單,如施加兩體或三體截斷,對于材料的力學性質描述較為準確,其研究尺度可達上百萬甚至千萬原子的體系[9]。但這樣的經驗力場并不適用于研究電子轉移、原子變價的化學反應過程,無法用于模擬含能材料燃燒與爆炸等復雜反應過程。反應力場是基于鍵級來描述體系的鍵合作用,鍵級隨著化學反應的發生自發進行調整[10],包括Airebo[11]、COMB3[12]和ReaxFF等力場[13-14]。其中,應用最廣泛的是Van Duin等[13-14]提出的ReaxFF力場,通過對鍵能、范德華能等多個能量項進行參數擬合來構建勢能面,目前在含能材料點火、燃燒、沖擊等方面均有大量研究[15-20]。利用反應力場進行分子模擬,結合模擬軌跡分析,可得到反應中間體、反應產物等隨時間的變化,從而提取其主要的基元反應、構建詳細反應機理。精確的力場模型是探究含能材料反應機理的基礎。此外,并不是所有含能材料體系均已開發ReaxFF力場,以AP為例,盡管已經廣泛驗證其作為氧化劑可以增加含能材料的能量釋放和反應活性,但是并未有前人開發相應的ReaxFF力場,因此目前并沒有關于AP燃燒過程的分子模擬研究。
第一性原理計算精度高,但計算成本大;分子動力學模擬效率較高,但是精度較低。兩種方法均無法兼顧精度與效率。如何高效率、高精度地重建勢能面是研究人員面臨已久的難題。近年來,基于機器學習的勢函數模型有望解決傳統計算方法的“卡脖子”問題。該方法繞過理論模型精確解釋的難題,通過完全數值化對抽象的物理規律和復雜的化學過程之間的潛在關系進行推斷,從而準確高效地構建反應體系的勢能面,實現多種復合含能體系的理論計算,建立分子結構與燃燒性能的聯系。
本文歸納了機器學習勢函數模型搭建方案及訓練集搭建策略,通過對高精度的量子化學勢能面進行近似,成功應用于含能材料燃燒爆炸方面的研究,包括常用的RDX、高氮炸藥CL-20、新型硝胺炸藥ICM-102、強氧化劑AP、高能顆粒(鋁和硼)等物質,并綜述了機器學習勢在碳氫燃料燃燒中的研究進展。最后總結了機器學習精度和效率方面的優勢,并對其發展趨勢提出展望。
1 機器學習勢
1.1 機器學習勢模型
精確構造含能材料的勢能面是開發機器學習勢的基本要求,也是研究其微觀燃燒機制的基礎。機器學習勢從第一性原理數據出發,用高維函數來描述分子結構與能量之間的關系,進而擬合勢能面,以較低的計算成本實現對目標體系的分子模擬計算。一個合格的機器學習勢模型需要至少滿足廣延性、對稱不變性和光滑性[9]。體系的廣延性是指體系總能量和原子數成正比,故機器學習勢的能量分解形式為:
(1)

(2)
為了達到上述要求,通常引入描述符D(Ri)的概念對原子三維坐標進行映射。對于進行等價對稱操作的原子坐標,其描述符相同,即:
D(Ri)=D(URi),?U∈u
(3)
從而將原子能量貢獻改寫為:
(4)
其中F為擬合方法。將原子坐標映射得到的結構描述符作為輸入,通過不同的擬合方法得到分子結構和能量的對應關系,即機器學習勢模型。模型的光滑性由擬合方法的光滑性和描述符共同保證。本節簡要介紹機器學習勢的擬合方法和描述符構造方法。
目前主流的機器學習擬合方法包括核方法和神經網絡方法。其中,核方法通過測量描述符與數據集中描述符的相似程度來獲得系統的能量或力。2010年,Csanyi等[21]基于核方法提出高斯近似勢(Gaussian Approximation Potential, GAP)模型,通過對原子化學環境相似性進行度量,采用高斯回歸過程對勢能面進行內插,從而得到原子能量、受力的最優估值[22]。分子構型越相似,在勢能面上的位置就越接近,構型的勢能也應當越相近[23]。這種方法并未預設函數形式,訓練過程人工干預較少,且可解釋性較強。目前在材料領域已經發表了大量的相關研究工作,如鈣鈦礦[24]、過渡態金屬[25-26]、硅烯[27]、半導體[28]、合金[29]等。2018年以來,基于核方法的(對稱)梯度域機器學習((Symmetric)Gradient Domain Machine Learning, GDML)[30]方法快速發展。與高斯近似勢不同,GDML模型在初始數據集中僅加入第一性原理計算的原子受力,通過對原子受力進行積分得到體系總能量[31]。引入受力項的模型比基于能量的模型自由度更高、參數更多、形式更靈活。
神經網絡勢函數(Neural Network Potential, NNP)模型是用一個多參數的非線性函數來擬合勢能面。早在1995年,Blank等[32]利用前饋神經網絡擬合勢能面,研究CO在Ni表面以及H2在Si表面的擴散問題。該模型的輸入坐標并不滿足平移、旋轉和交換對稱不變性,故前饋神經網絡僅僅應用于低維體系。2007年,Behler和Parrinello等[33]提出了高維神經網絡(High-Dimensional Neural Network, HDNN)模型, 采用以原子為中心的對稱函數矢量來描述原子的鄰域環境,每個原子用一個單獨的前饋神經網絡表示,從而預測每個原子對體系全局能量的貢獻。這樣的神經網絡預測能力更強,但需要反復調節和擬合大量的網絡參數, 且需要充足的訓練樣本。一般來說,神經網絡模型的深度比其寬度更受青睞,即隱藏層的層數比每層的節點數更重要[34]。這是因為更深的神經網絡具有更好的非線性表達能力,可學習更復雜的變化,從而擬合更復雜的特征。換言之,包含多個連續隱藏層的深度神經網絡的高維非線性函數擬合能力更為強大。2018年,鄂維南、張林峰等[35-36]基于深度神經網絡提出了深度勢(Deep Potential, DP)模型。同年,該團隊開發了開源工具DeePMD-kit[37]來支持DP模型的開發工作。目前DP模型已被用于材料、力學、化學、生物等各個領域的研究,如電池[38-39]、催化[40-41]、蛋白質[42-43]、含能材料[44-46]等。以DP模型為代表的機器學習勢模型,通過機器學習算法加速和大規模并行優化,可在保持第一性原理計算精度的同時實現上億原子的分子動力學模擬,將精確的物理建模帶入了更大尺度的材料分子模擬[47]。
機器學習勢通過構造結構和能量之間的映射關系來重建勢能面,因此采用何種描述符來提取分子結構特征進而用于訓練機器學習模型對于力場開發的精度和效率至關重要。Behler和Parrinello等[33]構造的原子中心對稱函數(Atom-Centered Symmetry Function, ACSF)滿足物理對稱性以及相對于移動原子的可微性,被廣泛集成于多個軟件中[48-49]。該方法以原子為中心,用徑向函數和角向函數將鄰域原子的化學信息編碼為復雜的“多體”形式,以完整表示原子的局域環境,并引入一個平滑函數,使其在截斷半徑處平滑過渡到零,以保證原子運動的平滑性和連續性。值得注意的是,ACSF方法給出的是構型的絕對描述,非常適用于在高維神經網絡勢中進行原子化學環境描述。
Bartok等[50]并未試圖建立局域構型的描述,而是直接建立兩個局域構型之間距離的對稱性度量,即平滑重疊原子位置(Smooth Overlap of Atomic Position, SOAP)。這種方法的主要依據是局域原子密度,將兩個局域構型之間的“距離”定義為兩個局域密度之間的交疊程度,并將核原子與其余原子的相對位置表示為徑向項和角向項的乘積。對于原子數量較多的團簇,SOAP描述符具有獨特的優勢,通常能得到更準確和穩定的勢能面。然而,SOAP給出的并非是某個構型的絕對描述,而是構型之間的距離,因此并不適合用于深度神經網絡,而非常適用于核方法[9]。同樣地,庫侖矩陣(Coulomb matrix)[51]描述并非針對局域原子環境,而是針對體系中所有的原子,將體系中N個原子的三維坐標轉換為一個N×N的矩陣,多用于開放邊界的體系,而非周期性體系的描述。庫倫矩陣保證了坐標的平移和旋轉不變性,對庫倫矩陣排序后可以保證交換對稱性。
描述符構造是開發機器學習勢的基礎,只有將描述符和擬合方法有效結合,才能最大程度地發揮機器學習勢的優勢。需要注意的是,關于何種分子描述符在機器學習勢模型中表現最優、從原子坐標中提取哪種特征,目前學界并無定論[8]。
1.2 數據集搭建方法
1.2.1 采樣方法
機器學習勢的開發過程中需要首先利用高精度的量子化學計算方法來構建體系的勢能面。從效率和精度的角度出發,通常采用領域中廣泛使用的密度泛函(DFT)方法。由此可見機器學習勢的精度上限是量子化學數據集的精度。考慮到DFT的計算成本,完全覆蓋復雜體系高維全局勢能面也是不切實際的,因此開發機器學習勢的核心是搭建合理的數據集。數據集的充分性、多樣性對于機器學習勢的精度至關重要。訓練集需要盡可能地覆蓋目標體系的勢能面,對不同含能材料組分、多種原子局域環境進行批量采樣,以得到精度高、拓展性強、普適性高的分子動力學機器學習勢函數模型。常用的勢能面采樣方法(見圖1)包括結構微擾法、分子動力學(MD)模擬方法和增強采樣方法。其中結構微擾法是對原子坐標施加結構縮放、錯位和隨機擾動,從而得到大量隨機分子結構;MD采樣方法采用現有的反應力場、經驗力場等進行分子動力學計算,得到分子的運動軌跡和反應路徑;增強采樣法則是通過人為施加約束,降低化學反應能壘,提高反應發生的概率。
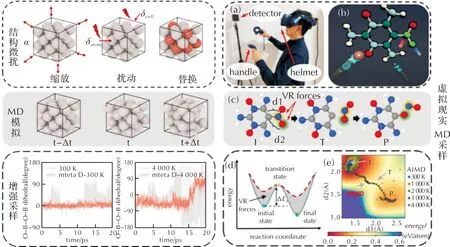
圖1 常見的采樣方法:結構微擾法、MD采樣法、增強采樣法和虛擬現實MD采樣法[45]
含能材料組分復雜,通常涉及到3種以上的化學組分,5種以上化學元素;且多應用于高溫(>3 000 K)、高壓(>10 GPa)等極端場景,其反應過程涉及到氣、液、固3個相態及兩相甚至多相之間的非均相反應,這些條件決定了含能材料機器學習勢的開發難度遠大于其他材料,往往需要更大規模的數據集來覆蓋可能存在的熱力學條件。因此在進行勢能面采樣時,需要有針對性地構造數據集。例如,在高溫條件下,分子化學鍵斷裂生成大量自由基,原子局域環境的復雜度顯著增加,因此初始數據集中需包括已知的反應路徑、過渡態結構和反應產物。為了描述含能材料的高壓構象空間,需要在數據集中引入不同壓力狀態的結構構象,即材料的狀態方程(壓力—體積曲線),以描述含能材料在極端場景的力學特性。
事實上,在傳統分子動力學模擬的時間尺度內,體系很難跨越較高的反應能壘,在相空間內的采樣效率具有明顯的局限性,使用增強采樣方法能夠更高效地對稀有事件(化學反應)實現采樣。然而,經典的增強采樣方法需要預先選定反應坐標,通過施加偏置勢或偏置力使得體系沿著既定的方向發生反應,考慮到含能材料復雜的分子結構和苛刻的熱力學條件,潛在反應坐標數量指數性增長,因此經典的增強采樣方法會存在反應坐標無法合理確定的困境,以至于對潛在分解反應路徑、過渡態結構等采樣不足。近年,本課題組[52]開發了一款虛擬現實(virtual reality, VR)分子動力學模擬軟件MANTA,為含能材料數據集的搭建提供了新的采樣工具。研究人員可以借助VR技術并憑借化學直覺對目標反應路徑開展實時交互,快速地對特定的反應過程勢能面進行采樣,將反應構型實時加入到訓練集中,是一種極具應用潛力的輔助采樣工具。總之,勢能面采樣是開發機器學習勢的關鍵步驟,在訓練過程中應結合上述手段,盡可能完備地描述化學反應的勢能面空間,以提高勢函數的準確性。
1.2.2 主動學習
主動學習是指在機器學習勢訓練過程中,對目標體系的相空間進行不斷地探索和補充,實現自動拓展數據集的一種訓練方法[53]。對于復雜體系,主動強化學習是一種十分合適的數據挖掘、構型探索、勢能面拓展方法。以DP-GEN[54]為例,這一工具是在DeePMD-kit的基礎上開發的同步學習工作流,其工作流程如圖2所示。
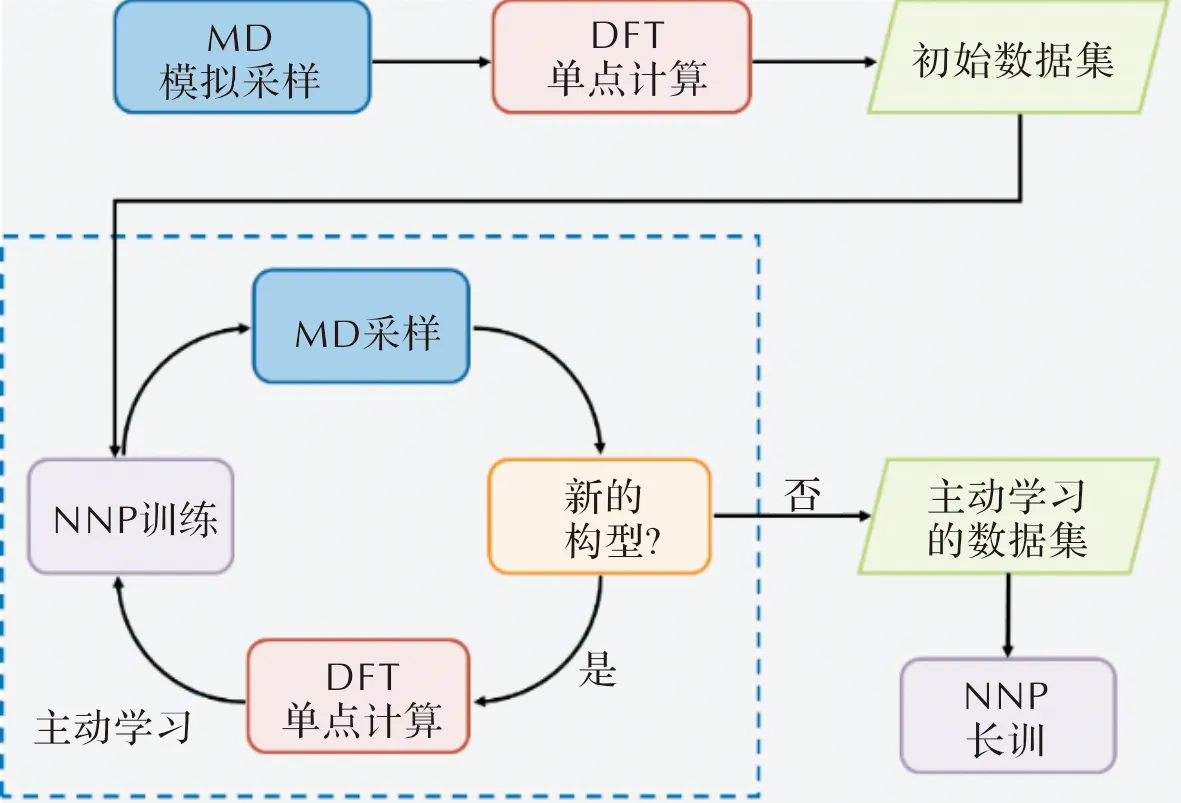
圖2 DP-GEN工作流程
針對初始數據集,調用DeePMD-kit工具,利用不同的神經網絡初始化設置,訓練多個機器學習勢函數模型。隨機選取其中一個勢函數模型進行分子動力學模擬(調用LAMMPS等[55]軟件),對運動軌跡中的構型能量和受力進行評估。若該模型預測構型的能量或受力與其他幾個機器學習勢函數模型有明顯偏差,則表示當前模型對于該構型的預測精度較差,需要補充到初始數據集中從而進一步優化勢函數模型。DP-GEN引入模型偏差來判斷多個機器學習勢間的誤差,通過設定誤差上下限來挑選候選構型。隨機挑選其中部分構型進行DFT計算得到新的數據,并加入到現有的數據集中重新訓練,從而得到精度比之前更高的機器學習勢模型。上述流程對數據集進行補充挖掘并得到新的機器學習勢模型,被稱為完成了一輪迭代。
采用DP-GEN方案進行多次迭代,每輪迭代可以改變系綜設置,完成在目標溫度、壓力或體積條件下的批量采樣和定量篩選,實現主動學習,旨在有效覆蓋所需樣本空間。高效的迭代訓練過程設計能讓勢能面覆蓋區域從局域空間平滑穩定地拓展到整個構象空間,例如從短的模擬時間逐步增加為長時間模擬,從低溫模擬逐步擴展為高溫模擬,從低壓模擬逐步增加為高壓模擬,從非反應體系過渡至反應體系。
2 基于機器學習勢的含能材料分子模擬
依據材料功能,含能材料組分一般可分為五大類,如圖3所示。

圖3 含能材料常見組分
其中,硝胺類是指以碳、氫、氧、氮等元素構建的具有爆炸性基團的化合物,是含能材料的主體部分[5]。硝胺類組分以環狀和籠狀的硝胺硝基化合物為主,如環三亞甲基三硝胺(RDX)、環四亞甲基四硝胺(HMX)、六硝基六氮雜異伍茲烷(CL-20)等。這類分子受限于反應產物的生成熱,導致其能量密度偏低。高能顆粒(鋁、硼、鎂等)憑借較高的燃燒焓被廣泛用做還原劑以提高含能材料配方體系的能量密度,調節其能量輸出特性,是強有力的動力來源和毀傷手段[56-58]。然而,高能顆粒的氧化層往往會限制其能量釋放速率,導致較長的點火延遲時間。在含能復合材料中加入聚偏二氟乙烯(PVDF)、高氯酸銨(AP)等強氧化劑有助于完全發揮納米顆粒的增益作用,其燃燒和熱分解性能極大改變了傳統含能材料的釋能機制[59-60]。為了降低含能材料感度,提高其力學性能,組分配方中通常包含少量黏結劑作為輔料。黏結劑組分多為高分子材料,如端羥基聚丁二烯(HTPB)、聚疊氮縮水甘油醚(GAP),這些高分子材料同樣包含一定含能基團以提高整體配方的能量密度。此外,近年來一部分高能量特性、熱穩定性、機械感度及力學性能優異的催化劑在含能材料領域中得到了廣泛的應用,如氧化石墨烯(GO)、銅(Cu)等[61-62]。
本章將介紹機器學習勢在含能材料分子模擬方面的研究進展,涉及到硝胺類化學物(RDX/CL-20/ICM-102)、氧化劑(AP)、金屬添加劑(Al/B)等組分的反應動力學研究。同時,包含了機器學習勢在碳氫燃料燃燒分子模擬中的進展。上述研究所采用的機器學習模型均為Zhang等[37]提出的DP模型,簡記為NNP。
2.1 硝胺類含能材料
2.1.1 RDX
黑索金(RDX)是一種典型的含能材料,穩定性好、爆炸威力大,被廣泛應用于火炸藥領域。Chu等[44]通過CP2K計算[63]將PBE/DZVP級別的第一性原理數據作為訓練集,基于DP策略開發機器學習勢模型對RDX熱解的反應動力學進行研究,并與ReaxFF[16]的結果進行對比。兩種方法得到的不同溫度下RDX分子數量變化趨勢相同,即體系分解速率均隨溫度升高而增加;但變化快慢卻大不相同,NNP模型預測的分解速率與第一性原理計算的結果一致,約為ReaxFF力場結果的兩倍。這表明開發的機器學習勢函數能準確描述RDX的熱解反應,而ReaxFF反應力場低估了分解反應的反應速率。此外,NNP模型和ReaxFF力場通過阿倫尼烏斯公式得到的RDX熱解反應活化能分別為105.6 kJ/mol和108.8 kJ/mol,與文獻值119.7 kJ/mol接近,即兩種力場模型均能較為準確地預測RDX分子發生單分子分解反應的活化能。
RDX熱分解的中間體和平衡產物主要包括NO2、NO、HNO2、N2、H2O、CO2和CO等。機器學習勢函數以第一性原理的精度成功復現文獻結果,即RDX受熱時N—NO2鍵會首先發生均相斷裂而生成NO2,中間產物NO2進一步發生反應生成NO和H2O分子。當達到平衡濃度時,體系中N2含量最多,其次是H2O和CO2。與ReaxFF模型的結果相比,機器學習勢預測到更多的NO分子生成量、更快的H2O生成率以及更高的N2平衡濃度,與文獻結果一致[64]。通過對分子動力學模擬的軌跡進行分析可以得到RDX熱解的詳細反應路徑,如圖4所示。RDX的分解機制可分為兩個階段:(1)N—N鍵斷裂生成NO2分子和RDR中間體;(2)中間體RDR進一步發生N—N鍵斷裂、開環反應、或氫加成反應。其中,關鍵中間體NO2發生氫加成反應會生成HONO,進而生成NO、N2、OH和H2O等物質。

圖4 RDX分解的中間產物及詳細反應路徑[44]
近鄰分子間的相互作用在很大程度上會影響甚至改變含能材料的分解路徑[6]。不同密度的RDX分子和RDR中間體發生N—N均裂、氫提取和開環反應的通量并不相同。在高密度情況下,由于空間位阻效應,近鄰分子會阻礙RDX分子及其中間體的開環反應;隨著密度降低,近鄰分子的影響減弱,開環反應通量增加,氫提取反應通量減少,RDX的熱解機制與孤立分子的第一性原理結果相近。上述研究結果在保持第一性原理計算精度的同時,從原子尺度闡明了RDX的反應路徑,建立了RDX熱分解詳細反應動力學機制。
2.1.2 CL-20
第三代高爆炸藥CL-20是能量和密度最高的單質炸藥之一,在氧平衡、爆速、爆壓和堆積密度等方面均優于其他含能材料。然而,對機械刺激的高敏感性限制了其應用前景。近年來,共晶技術在含能材料領域已成為一種廣泛應用的策略,以更好地平衡感度和能量輸出之間的關系。現有的實驗和理論工作表明,CL-20與其他含能材料共晶結合可以提高沖擊穩定性和熱穩定性。盡管已經有許多關于CL-20的共晶材料被設計出來,但提高穩定性的具體微觀機理仍有待進一步揭示。
Cao等[46]構建了β-CL-20和CL-20/TNT共晶體系PBE/DZVP級別的神經網絡勢能函數,并在此基礎上開展分子模擬研究以揭示CL-20/TNT共晶穩定性增強背后的分子機制。為了深入探索β-CL-20和CL-20/TNT共晶的熱解過程,基于機器學習勢進行了分子動力學升溫模擬。對于β-CL-20體系,CL-20分子在溫度升高到大約1 200 K時開始分解;而在CL-20/TNT共晶體系中,CL-20分子發生分解的溫度高達1 400 K左右。通過比較起爆溫度,可以發現共晶CL-20/TNT的熱穩定性明顯提高。在不同的升溫速率下,也能觀察到相似的結果,即在CL-20/TNT共晶體系中的起爆溫度總比在β-CL-20體系中的要高。這是由于共晶體系會穩定生成分子間氫鍵,而在單質體系中并沒有氫鍵的出現。分子間的氫鍵相互作用可以有效延遲CL-20分子的裂解,降低活性物種的反應速率,這一結果與張朝陽等[65]對含能材料晶體結構的研究結果一致。通過對分子動力學模擬軌跡進行分析,得到單質CL-20分子主要的熱分解路徑,如圖5所示。與之前的研究一致,CL-20分子熱解以N—NO2鍵的斷裂為主,脫去NO2后的中間體進一步發生N—N斷鍵和C—C斷鍵,生成產物NO2和CxNy小分子。在共晶體系中,也觀察到了類似的初始熱解路徑,但是速率較低。TNT中的自由基與游離的NO2分子會發生進一步反應,如TNT中的氫自由基與游離的NO2分子結合生成HONO,TNT中的甲基自由基與NO2結合生成甲醛(HCHO)和NO。由于TNT中的C—N鍵和六元環相對穩定,因此在模擬過程中很難發生NO2的分離和開環反應。值得注意的是,從CL-20中分離出的NO2與TNT的反應進一步減緩了初始階段NO2的生成速度,阻礙了CL-20分子的進一步分解。因此,TNT可以被視為緩沖碎片,以降低活性中間體與CL-20分子碰撞的概率。上述工作從原子尺度揭示了CL-20/TNT共晶體穩定性增強背后的物理化學機制。

圖5 CL-20熱解的初始反應路徑[46]
2.1.3 ICM-102
新型含能材料的設計是提升我國先進武器裝備綜合性能的有力支撐。張慶華等[66]將“材料基因組工程”的研發模式引入到了含能材料領域,針對含能材料能量與安全匹配性差的問題,運用計算機輔助分子高通量設計與篩選的手段,成功研發了一種高能鈍感炸藥分子ICM-102(C4H6N6O4)。其能量水平高于低感炸藥LLM-105,感度與鈍感炸藥TATB相當[67],實現了炸藥能量與安全性的平衡。Chu等[45]基于虛擬現實增強采樣(VRMD)方案構建了PBE/DZVP級別的數據集,并開發機器學習力場來描述ICM-102的反應動力學。其中,第一性原理計算采樣的構型與初始構型接近,隨溫度的升高,構型在勢能面上的分布區域擴大,但主要集中于低能區域,導致反應構型數量有限,不能完整描述整個反應過程。而VRMD方案通過手柄施加外力,并依靠化學直覺對經驗的反應位點、反應路徑進行實時交互采樣(見圖2)來構建數據集,可以直接對斷鍵反應等稀有事件進行直接采樣,有效提高了高能壘反應過程的采樣效率。
數據集的質量對于機器學習模型的性能至關重要。通過主成分分析可提取采樣結構的主要特征并進行數據降維,從而將采樣到的結構劃分為晶體構型、部分分解構型和氣態產物,如圖6所示。沿著總反應發生的方向,即主成分PC1增加的方向,兩種采樣方案得到的構型質量均逐漸減少,但是對應的數密度卻明顯不同。由于反應能壘的存在,直接分子動力學采樣的數據集范圍明顯集中在低能勢阱,在高能區域的采樣較少,即得到的構型大多并未涉及到反應的發生。而VRMD采樣方案可針對能壘較高的反應過程進行采樣,作為AIMD采樣方式的補充,極大地拓展了勢能面的采樣空間。此外,單一的AIMD采樣方案得到的模型能量精度為0.014 eV/atom,兩種方法相結合得到的模型偏差僅有0.006 eV/atom。這表明VRMD采樣方案可以作為補充手段,來構造更為均衡、更豐富的訓練集,從而得到更精準、更有效的勢函數模型。

圖6 AIMD和VRMD采樣方案的主成分分析[45]
通過高溫分子模擬,Chu等[45]發現ICM-102熱分解的主要中間體和產物包括C6H5N6O4、H2O、NO、N2、CHNO和CO2。利用聚類分析的方法可以將所有出現過的分子構型分為八類:前兩類分子表示ICM-102及其失去一個小分子基團后的產物;第三、四類分子表示ICM-102分子經過開環反應的產物;第五、六、七類分子表示開環反應后進一步生成的含碳或含氮的分子;第八類則是反應過程中的水、一氧化氮和氮氣等小分子氣體。以熱解反應網絡為基礎,可直接依據通量分析推演任意中間物質的詳細反應網絡(見圖7)。
2.2 AP
AP的有效氧含量高、機械感度低、相容性好,爆炸過程時產生的氣體多、無固體殘渣、成本低,是固體推進劑和火炸藥等最重要的強氧化劑[68-69]。AP的熱分解是一個復雜的物理化學過程,涉及到吸熱相變和放熱反應。目前AP的熱分解研究以傳統實驗為主,如使用熱重分析(TGA)、差示掃描量熱法(DSC)和質譜分析(MS)等來確定分解速率和中間產物。傳統的實驗手段并不能在極小的時間、空間尺度上直接觀測到AP復雜的反應機制。而AP反應力場的缺失制約了研究者對其熱分解機理的微觀研究,是火炸藥領域中的“卡脖子問題”。Chu等[70]基于第一性原理計算(PBE/DZVP級別)首次建立了AP的機器學習勢,對其熱分解過程進行分子動力學模擬,從原子尺度成功提取AP分解的動力學演化過程,填補了目前含能材料燃燒機理缺失的重要部分。通過分子模擬復現TGA/DSC實驗,將AP晶體從常溫加熱至3 000 K,所開發的機器學習勢可描述AP熱分解過程中的質量變化和熱流速率,即從正交相到立方相的吸熱相變,以及隨后的放熱分解反應。
AP分子由NH4和ClO4基團構成,通過直接分子動力學模擬,機器學習勢模型預測其分解過程中生成的中間體和反應產物多達三萬種。其中,反應的中間體主要是NH3、ClO2、ClO3、HClO4。隨著反應的進行,最后的平衡產物以H2O、NO、O2、Cl2、N2和HCl為主。通過提取熱分解過程中的主要物質和生成路徑得到AP反應網絡,如圖8所示。

圖8 AP分解的產物數量變化和主要反應路徑[70]
考慮到AP中的強氧化性元素較多,據此可以將整個分解動力學劃分為AP的初始分解、氫化學、氮化學和氯化學。初始分解過程以質子轉移為主,NH4中的氫自由基會轉移到HClO4中并生成NH3,或與ClO4中的O自由基結合生成ClO3和OH。在初級分解之后,氮化學、氫化學和氯化學反應同時進行。特別指出的是,上述分子模擬預測到一些實驗未曾報道過的雙分子反應,例如NH4+NH2→2NH3和ClO4+NH3→HClO4+NH2。這些氣相物種和AP之間的雙分子反應可能會影響AP的分解過程。上述研究以第一性原理計算的精度首次揭示了AP的詳細反應機制,克服了AP反應力場缺失對基礎研究的制約,為建立復合含能體系的反應動力學模型提供了新的機遇。
2.3 高能顆粒(Al、B)
金屬顆粒能量密度大、燃燒熱值高,是極具吸引力的含能材料組分,能顯著提高固沖發動機的性能,增大導彈武器的射程,提高戰術武器的作戰效能和毀傷效益[40, 71-72]。金屬作為燃料和燃料添加劑可以減少燃燒不穩定性、提高燃燒速率、降低爆震敏感性[57, 73]。其中應用最廣的金屬顆粒是鋁,其燃燒熱值高、能量密度大且儲量大,是重要的含能材料添加劑。近期本課題組基于PBE/DZVP級別的第一性原理數據建立機器學習勢模型,訓練集中包括Al、Al2O3、界面以及氧氣擴散等數據。所開發的模型可以復現晶體鋁的狀態方程曲線(見圖9),計算結果與DFT預測基本一致。模型在極端的壓縮和拉伸情況下仍然保持著良好的性能表現,因此可以用于研究鋁在沖擊波下的力學響應行為。值得注意的是,盡管訓練集中僅包含了單位原子體積在10~25 ?3/atom的數據(圖中虛線區域的數據),機器學習勢模型可以準確地預測7~32 ?3/atom范圍內的狀態方程,這證明了機器學習勢函數具有一定的外推性能。

圖9 鋁的狀態方程曲線
在所有金屬和類金屬中,除有毒性的鈹外,硼的體積熱值和質量熱值最高,分別為138 kJ/cm3和59 kJ/g[74-75]。盡管硼的理論燃燒性能優異,但是硼的熔點(2 348 K)和沸點(4 043 K)都很高,導致顆粒難以點火,限制了硼粉的實際應用。此前Liu等[76]、Wang等[77]采用分子模擬的方式研究了硼顆粒的微觀燃燒機理,然而這些工作使用的勢函數是基于氨硼烷[78]開發的,并未針對硼燃燒進行優化。這里我們提出一個問題:一個力場模型的開發數據集中并不包括硼單質的數據,這樣的模型可直接用于硼燃燒的模型嗎?勢函數模型是針對目標體系特定的應用場景而開發的。一般而言,訓練集只是全局勢能面上的局部空間,在勢能面上的外推性能非常局限。例如,同樣含有C/H/O/N元素的反應力場可以描述RDX的熱解反應,卻不能直接用于CL-20的模擬,會導致預測誤差迅速升高。因此,在進行分子動力學模擬之前,需要對勢函數開發的訓練集進行分析,確保數據集覆蓋了關注的勢能面區域,避免超出訓練集的外推空間,并進行物理化學參數驗證,如晶格參數、狀態方程、融點、比熱容等。
Chang等[79]利用CP2K構建了PBE/DZVP級別的量子化學數據集,開發了用于描述硼和硼氧化合物的DP模型。開發的DP模型對能量、受力、狀態方程、晶格參數和徑向分布函數等進行驗證,機器學習力場的精度均和第一性原理計算一致。相比之下,針對氨硼烷燃燒開發的ReaxFF力場預測結果非常差[78]。例如,文獻中晶體硼的單位原子體積為7.25 ?3/atom,機器學習預測值為7.01 ?3/atom,而ReaxFF力場的預測值高達12.58 ?3/atom,顯著高估了晶體硼的平衡結構。以上這種明顯誤差主要來源于ReaxFF力場的濫用,考慮到上述工作的ReaxFF是針對氨硼烷燃燒體系,在其開發過程中不含任何硼單質晶體結構,因此這套ReaxFF力場參數無法支持硼燃燒的分子動力學模擬。另外,這直接表明研究人員應當避免任意拓展ReaxFF力場的應用范圍。
Chang等[79]采用DP模型模擬了納米硼顆粒的融化行為。通過計算顆粒全局Lindemann指數和勢能變化,得到不同粒徑硼顆粒的熔點,歸一化后的熔點如圖10所示。對于3 nm硼顆粒,其融點高達1 992 K,而同樣粒徑的鋁鎂顆粒在該溫度下已完全融化并蒸發到氣相環境中[80-81]。這表明納米硼顆粒的燃燒機制仍然是以表面反應為主。通過分析原子Lindemann系數演化,作者發現硼融化的過程包括初始加熱、表面預融、內核預融、完全融化和繼續升溫等五個階段。該融化過程遵循Nanda等[82]提出的液核生成假設,即表面先熔化隨著溫度逐漸向內核區域擴散。上述融化機制表明,在達到融點之前,表面預融的硼顆粒會和周圍的顆粒發生燒結、團聚,阻礙氧化劑向顆粒內部擴散,從而導致點火延遲。基于機器學習勢的融點研究從原子角度解釋了硼顆粒在燃燒爆炸應用時受限的物理化學機制。

圖10 鋁、鎂、硼的歸一化融點[79]
2.4 碳氫燃料
碳氫燃料具有高能量密度和低成本等特性,是火箭推進劑、噴射推進劑等航空燃油的主要組成部分,其反應機制是燃燒反應動力學中重要的研究對象。Zeng等[83]將深度勢模型引入燃燒領域研究,基于MN15/6-31G**級別的第一性原理數據開發機器學習勢,對甲烷氧化的基本反應機理和動力學過程開展研究。分子模擬結果表明,甲醛(CH2O)和甲氧基(CH3O)是最關鍵的中間產物,主要反應路徑為:CH4→CH3→CH3O→CH2OH→H2CO→HCO→CO。甲烷燃燒起始反應是氧氣的抽氫反應,生成甲基(CH3)和HOO自由基。隨著反應進行,不斷生成的OH、H和HOO自由基會與甲烷發生抽氫反應,生成大量的甲基。甲基進一步被氧化為甲醛,接著失去一個氫原子形成甲酰基(HCO—),隨后可以通過碰撞解離,與氧分子反應失去氫原子,從而形成一氧化碳(CO)。上述反應路徑與文獻結果一致,以第一性原理的精度搭建了甲烷燃燒的反應網絡。
高溫下碳氫燃料的不完全燃燒是碳煙前驅體多環芳香烴(Polycyclic aromatic hydrocarbons,PAH)的重要來源。碳煙的初生受燃料結構、氧氣濃度、溫度等因素的影響,同時存在物理和化學機制的復雜過程。Wang等[84]通過Gaussian計算得到MN15/6-31G**級別的數據集從而搭建機器學習勢函數模型,并根據化學經驗設計反應物,對A1~A9多環芳烴化合物(PAH)生成路徑進行模擬研究。以單環和雙環PAH的生成過程為例,機器學習勢函數成功預測到文獻中丁二烯和乙炔反應生成苯環、五元環碳碳鍵斷裂生成萘的過程。在生成三環芳香烴化合物的模擬中,觀測到的主要反應為經典的抽氫-乙炔加成(HACA)反應,即乙炔與1-萘結合生成苊并致使五元環閉合。此外,還發現一些新的反應路徑,如苯基和C4H4反應生成一條長碳鏈,然后通過閉環和抽氫反應生成茚。這些新的反應路徑對于構建詳細的碳煙生成模型具有重要意義。
Chu等[85]基于DP方案,發展了PBE/DZVP精度級別的機器學習勢函數,在此基礎上進一步建立了納米反應器算法以加速反應、探究密度對于反應路徑的影響。納米反應器用一個球形壓頭周期性地壓縮和放松體系,使體系的密度發生變化,從而改變分子碰撞頻率,加快反應進程。此外,高溫手段使得分子動能增加,運動更為劇烈,同樣可以顯著加快反應進程。圖11以乙炔體系多聚化反應為例,對比了常規分子動力學計算結果和納米反應器算法加速的計算結果。

圖11 3種分子動力學模擬過程中乙炔分子數量變化及物種演化[85]
其中,方案一表示納米反應器中的反應進程,方案二和方案三分別為1 500 K和3 000 K下傳統的NVT模擬。值得注意的是,納米反應器算法可以顯著加快反應進行,其反應速率是同樣溫度MD結果的50倍。納米反應器中的乙炔分子數量變化與1 500 K下縮放的結果趨勢相同,其反應途徑相同,僅僅存在乙炔的聚合反應。這表明,增加濃度或壓力只影響碰撞頻率,而不影響反應路徑。高溫會影響體系的總能量,進而改變反應進程,因此方案三中的反應路徑與前兩種方案的結果并不相同,涉及到氫抽離和氫加成反應。上述結果表明納米反應器在不影響反應路徑的前提下提高了碰撞頻率,為研究成核和低溫反應等緩慢動力學過程的反應機理提供了一種很有前景的工具。
作者進一步采用納米反應器方法來加速碳煙的成核過程,考慮了文獻中報道的7種不同PAH分子的成核反應進程。圖12展示了從納米反應器中的不同前體收集的最終產物。對于小分子前驅體,最終生成的多環芳香烴以氣態形式排列。大多數自由基會形成交聯的二聚體或三聚體,并未觀察到大的團簇。質量較大的前驅體分子最終會在納米反應器的中心形成團簇。而對于大型前驅體,其最終產物呈球形堆積結構,不同層之間存在交聯。上述研究表明,碳煙的最終產物形態與前驅體的大小有很大關系。隨著前驅體尺寸的增加,產物顆粒從無序的團簇變成了有序的緊密堆積結構。

圖12 不同前驅體在納米反應器中生成的最終產物[85]
3 機器學習勢的計算優勢
原子間相互作用勢的函數形式、理論假設和近似合理性決定了其精確度,進而影響分子動力學模擬結果的可靠性[86]。一般的力場假設的函數形式較為簡單,如施加兩體或三體截斷。目前含能材料分子動力學研究常采用的ReaxFF力場,是僅對能量項進行擬合,并未考慮原子受力項,其函數形式復雜,并行效率低。機器學習勢是利用其底層算法對高維函數的強大擬合能力,以一種基于數據驅動的方式近似第一性原理原子相互作用。圖13總結了目前含能材料和碳氫燃料中的機器學習勢模型精度。其中,Zeng等[83]開發的甲烷燃燒機器學習勢是基于Gaussian計算得到MN15/6-311G**精度的第一性原理數據,其余機器學習勢的數據集均為CP2K計算的PBE/DZVP級別精度。

圖13 NNP勢和ReaxFF反應力場精度對比
總體來說,各個體系中的機器學習勢表現均優于ReaxFF力場,能量和受力的預測精度分別提高3.5倍和6.6倍(RDX、Al和碳煙體系)。最初的ReaxFF力場開發是針對碳氫燃燒并逐步拓展到CHON類含能材料研究,經過近二十年的驗證和優化,其參數精度較高、力場表現較好。從圖13中可以看出,經過DP-GEN迭代的RDX和碳氫的勢函數模型精度都僅僅是略高于ReaxFF。以RDX為例,在能量預測方面,NNP力場相較于ReaxFF力場能量精度提高4倍,受力精度提高9倍。這是由于ReaxFF擬合過程僅考慮了能量項,并未考慮受力項,所以對于原子受力的偏差較大。值得注意的是,對于反應力場訓練集和目標體系并不匹配的力場而言,ReaxFF的表現不盡人意。以硼的機器學習勢為例,ReaxFF力場的能量和受力偏差分別是NNP模型的180倍和18倍。這樣顯著的偏差強調了力場訓練集對于目標體系的適配性和移植性。此外,勢能面的采樣質量對于機器學習勢的精度也至關重要。對于新型含能材料ICM-102,VRMD采樣方案可對勢能面上的高能區域進行采樣,從而提升數據集的整體質量。采用VRMD采樣方案使得能量預測偏差降低58%,受力預測偏差降低37%,這種輔助采樣策略極大地提高了模型的整體精度。
ReaxFF力場在擬合過程中未考慮材料在極端條件下發生壓縮、膨脹等力學行為。在強烈的爆炸沖擊作用下,含能材料的溫度會瞬間急速上升,壓力升高至幾十GPa,體積減少30%[87]。機器學習勢可以在訓練集中加入不同壓縮和拉伸條件下的高溫高壓數據集,這對于研究含能材料的沖擊壓縮效應具有現實意義。圖14為不同計算方法得到的RDX晶體狀態方程。其中,機器學習勢預測結果與第一性原理計算結果一致。

圖14 RDX晶體的狀態方程曲線[44]
值得注意的是,機器學習勢具有一定的外推性能。在圖14中,機器學習勢的訓練集僅考慮了0.9~1.0的壓縮比,即圖中的矩形部分。但是該模型能成功預測0.8~0.9區間的狀態方程曲線。相比之下,反應力場由于并未考慮勢能面上的壓縮區域,無法復現RDX的狀態方程曲線,預測的平衡結構體積偏大,在進行弛豫后將無法得到準確的晶體構型。此外,反應力場明顯高估了壓縮過程中的勢能,在爆炸或沖擊載荷的研究中可能會人為形成局部熱點,加速反應的發生。因此,目前的反應力場的精度并不能滿足爆炸沖擊下材料性能的研究需求。Chu等[44]提出的機器學習力場更能準確描述RDX晶體在燃燒爆炸等極端環境下的理化性質和分解過程。
除了擁有更高的計算精度,機器學習勢計算效率也更為優異。圖15以RDX晶體為例對比了不同方法的計算效率,體系規模從100個至10萬個原子。機器學習勢在保持DFT精度的基礎上,計算效率與體系規模呈線性關系,約為反應力場的27倍,比DFT效率提升了4個數量級。這是由于機器學習勢模型計算是基于GPU硬件實現的。對于RDX晶體而言,機器學習勢的計算成本與體系規模呈O(N)的趨勢。而對于密度較低的氣相碳反應體系,機器學習勢模型的計算成本與體系規模呈O(NlogN)的趨勢。機器學習勢的優異表現使得用第一性精度探索具有數十萬甚至數千萬原子系統成為可能,可以從原子角度研究含能材料的復雜反應機制,為建立詳細的含能材料反應動力學模型提供了新的機遇。

圖15 密度泛函理論(DFT)、反應力場(ReaxFF)和機器學習勢(NNP)的計算效率對比[44]
4 挑戰與展望
分子動力學模擬能夠在原子尺度上揭示含能材料的微觀化學反應路徑,為研究復雜體系的反應動力學機理提供了一條新的途徑。分子動力學模擬的關鍵是力場參數,它能夠描述反應的勢能面結構,決定了模擬的精度和效率。從頭算分子動力學準而不快,反應力場計算快而不準,機器學習勢兼顧計算精度和計算效率,在含能材料燃燒與爆炸領域得到廣泛應用。
機器學習算法不需要預設物理化學規律,可直接從計算數據出發,準確推斷函數關系。得益于這一優點,機器學習勢往往能夠重現含能材料反應過程勢能面變化,進而研究其熱解或燃燒過程的詳細反應機理。同時,數據驅動這一特性也給機器學習勢的應用帶來了一系列的挑戰。機器學習勢的開發需要大量高質量數據,且很難先驗地判斷數據集的完備性。雖然,數據集的規模可能很大(大于10萬量子化學計算數據),但它并不足以對目標體系進行充分采樣。例如,基于低溫分子模擬采樣搭建的數據集,其機器學習勢無法準確外推到高溫體系。因此,數據集需要盡可能地包括目標體系的整個勢能空間。其次,訓練集一般是基于第一性原理計算開發,而第一性原理計算本身也存在一定誤差,受到基組大小、泛函精度等因素的限制。通過提高第一性原理計算的理論方法可以減少計算誤差,然而也會極大地增加計算耗時,尤其是考慮到現有的勢函數開發往往需要十萬量級的第一性原理計算數據,搭建高精度數據集的成本極其昂貴。未來含能材料的機器學習勢函數開發面臨著如下挑戰:
一是如何對極端條件下復雜反應勢能面進行充分采樣。此前的工作主要針對單質體系,在實際應用中含能材料的配方日益復雜,包含硝胺類材料、高分子化合物、金屬材料等多種組分。隨著元素種類的增加,反應空間構型的數量呈現指數級增長,需要包含更多的反應路徑、中間體構型。這對如何有效地構建訓練集提出了新的要求。目前存在主動學習、勢能面掃描等思路。Zhang等[54]提出了DP-GEN主動學習算法,通過比較不同模型的不確定性動態生成新的數據點,從而提高模型對全局勢能面的覆蓋度。另一方面,研究者可以根據對化學反應的認知,設計勢能面掃描算法對化學反應路徑上的結構構型進行采樣。Liu等[88]提出采用隨機勢能面行走方法(SSW),利用化學鍵振動對構型施加微擾,從而高效搜索復雜結構的全局勢能面。Zhang等[89]提出了一種準經典軌跡法構建局部勢能面的方法,可針對特定反應開發高精度勢能面。增強采樣方法也是一種高效采樣方法,可以作為常規采樣方法的重要補充。本課題組最近結合虛擬現實技術開發了一種增強采樣工作流[52],在虛擬現實空間中直接對關鍵反應路徑的勢能面進行采樣。和傳統的分子動力學模擬直接采樣相比,上述幾種方法都能夠提高勢能面采樣的效率,克服勢能面上存在的高勢壘,更好地描述復雜反應過程。值得注意的是,含能材料應用中常涉及極端的高溫高壓環境,在構建訓練數據集時需要把極端條件的構型納入考慮。
二是如何提高機器學習勢訓練集的理論精度。機器學習勢的精度依賴于訓練集選取的量子化學計算方法,特別是特定的物理化學性質或反應體系對DFT的泛函精度要求較高(如反應能壘、生成焓等),需要使用高精度的雜化甚至雙雜化泛函計算方法。然而高精度的DFT計算成本隨著系統規模的擴大而急劇增加,這限制了高精度DFT數據的數量,難以滿足機器學習勢開發的需求。Chen等[90]提出了DeePKS方法,通過機器學習方法開發DFT泛函,從而極大地縮減了高精度DFT計算的耗時,有望實現大規模的高精度DFT訓練集構建。Smith等[91]提出了一種程序框架實現了小樣本數據的機器學習勢函數開發。他們先使用普通的DFT數據訓練模型,然后使用遷移學習方法在高精度的DFT數據上對模型進行再訓練,從而獲得高精度的機器學習勢函數。在未來開發含能材料的機器學習勢函數時,結合上述方法有希望更好地構建高精度DFT數據集。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
中國外匯(2019年17期)2019-11-16 09:31:14
光學精密工程(2016年6期)2016-11-07 09:07:19
核科學與工程(2015年4期)2015-09-26 11:59:03
現代企業(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
中國火炬(2010年7期)2010-07-25 10:26:09

- 火炸藥學報的其它文章
- 計算含能材料專刊序言
- 五類新型雙環四唑含能化合物的設計與性能研究
- Pyrrole and Pyrimidine Derivatives as Possible Electron Donors for Colored Charge-Transfer Complexes with a Weakly Electrophilic Energetic Material, FOX-7: A Theoretical Study
- 4,4′-偶氮-1,2,4三唑與4,4′-聯-1,2,4-三唑含能性質的DFT研究
- 損傷累積下PBX炸藥低速撞擊點火行為的數值模擬
- 高能鈍感金屬配合物的新設計策略:集全四唑高氮配體、少硝基和氧平衡接近于零為一體