基于全基因組重測序分析新疆細毛羊遺傳多樣性
2023-06-20 05:13:00陳開旭郭翠潔任斐兒李曉斌劉武軍
新疆農業科學 2023年5期
關鍵詞:新疆
陳開旭,郭翠潔,楊 帆,任斐兒,李曉斌,劉武軍
(新疆農業大學動物科學學院,烏魯木齊 830052)
0 引 言
【研究意義】我國羊毛生產主要集中在新疆、內蒙古、甘肅、青海、吉林等省(自治區),為我國目前細毛羊的主產區,占全國細毛羊總產量的97%[1],對畜牧業經濟發展有重要影響[2]。新疆細毛羊是毛、肉兼用細毛羊品種,由高加索細毛羊公羊與哈薩克母羊、泊列考斯公羊與蒙古羊母羊進行雜交培育而成[3]。該品種適于干燥寒冷高原地區飼養,采食性好,生活力強,耐粗飼料等,利用全基因組重測序技術分析新疆細毛羊的遺傳多樣性,對發掘新疆細毛羊的遺傳資源,保護其多樣性基因庫有重要意義。【前人研究進展】新疆綿羊品種較多[4],新疆細毛羊具有適應性強、毛質好、肉質多、產毛量高、凈毛率高、體大、耐粗飼、遺傳性能穩定等優良品質。開發和利用新疆細毛羊這一寶貴遺傳資源,對我國畜牧業的發展具有重要意義[5]。遺傳多樣性的本質是地球上物種發生、進化和變異的基礎[6,7],遺傳多樣性的研究方法已從傳統的形態標記、染色體標記及生化標記等發展到分子水平遺傳標記的研究[8]。【本研究切入點】目前有關基于全基因組重測序分析新疆細毛羊遺傳多樣性的研究文獻較少,需利用新疆細毛羊的全基因組重測序數據,檢測不同綿羊品種的核苷酸多態性和單倍型多態性。【擬解決的關鍵問題】通過全基因組重測序的方法尋找新疆細毛羊的高密度 SNPs 遺傳標記,運用雜合度、連續純合子區域、連鎖不平衡分析評估新疆細毛羊的遺傳多樣性。
1 材料與方法
1.1 材 料
1.1.1 細毛羊
從伊犁鞏乃斯種羊場隨機選取10只新疆細毛羊(Xinjiang Fine Wool Sheep,XFW),通過頸靜脈采血法采集10mL頸靜脈血于含有抗凝劑(乙二胺四乙酸(EDTA)鹽)的真空采血管中,置于-80℃冰箱中凍存備用。10只巴音布魯克羊(Bayinbuluke Sheep,BYK)、10只策勒黑羊(Cele Black Sheep,CLE)、10只阿勒泰羊(Altay Sheep,ALT)的基因組數據下載自 NCBI 數據庫(GenBank檢索號SRP363313)的全基因組重測序數據。
1.1.2 DNA 提取與質量檢測
按照血液全基因組DNA提取試劑盒(QIAGEN)操作步驟,從新疆細毛羊全血樣品中提取基因組DNA。DNA樣品的濃度和純度(DNA樣品中蛋白質和RNA等污染),通過NanoDrop 1000超微量紫外分光光度計進行檢測,通過凝膠電泳試驗判斷,DNA樣品的完整性和降解,綜合評判DNA樣品的質量。
1.1.3 全基因組重測序與序列
1.1.3.1 測序文庫構建
全基因組重測序對基因組DNA的質量要求標準為:基因組DNA經瓊脂糖凝膠電泳檢測結果顯示:條帶單一且清晰明亮,無拖尾(降解)現象,基因組DNA經濃度測定結果顯示:濃度≥ 50ng/μL,總量大于3 μg,OD260/OD280=1.8~2.0(無蛋白及RNA污染),則認為該DNA樣品質量符合測序要求。
測序文庫構建:(1)用超聲波將檢測合格的基因組DNA樣品隨機打斷成長度約為500 bp的片段;(2)用末端修復酶修復DNA,以提高DNA連接入載體的效率;(3)片段3’ 末端連接多個堿基A尾;(4)連接測序接頭;(5)對DNA片段進行選擇;(6)PCR擴增DNA片段;(7)再次進行DNA片段的選擇和純化。
文庫構建完成后使用熒光計(Qutit2.0)進行初定量,根據定量結果將DNA片段濃度稀釋至1 ng/μL。使用Agilent 2100 bioanalyzer對Insert Size檢測,確保Insert Size符合文庫要求。使用Q-PCR方法準確定量文庫的有效濃度。利用Illumina Hiseq 4000高通量測序平臺對文庫進行雙末端全基因組重測序。
1.1.3.2 測序質量評估與過濾
對完成全基因組重測序后的基因組序列數據進行質量評估與過濾,包括:去除無法配對的讀長(reads)和低質量的reads、去除reads兩端的barcoding序列 和index序列。數據清理后,獲得10個文庫的20個.fastq文件。采用FastQC 軟件對測序后的基因組序列質量進行統計,內容包括:綿羊基因組測序序列reads中C、T、G、A四種堿基的含量和偏好性;基因組序列中的reads數目和長度的總體分布;reads中每個堿基的總體質量。
1.1.3.3 全基因組序列比對及比對文件預處理
下載綿羊參考基因組序列文件(Oar_4.0)(https://www.ncbi.nlm.nih.gov/assembly/GCF_002742125.1)并建立其索引文件。綿羊參考基因組作為比對模板,使用 BWA MEM[9]程序對每個綿羊個體基因組文庫測序生成的2個.fastq文件分別進行比對,生成2個.sam文件。10個DNA文庫,共生成20個.sam文件。將下載的10只策勒黑羊、10只巴音布魯克羊、10只阿勒泰羊個體的測序文件按照同樣的方法比對,生成60個.sam 文件。整理和統計比對后的reads數量、比例、reads的配對率等結果。
1.1.3.4 遺傳變異的鑒定、過濾和注釋
依據與參考基因組比對得到的結果,使用SAMtools[10]和GATK(版本號:3.6-0-g89b7209)[11]兩款軟件包分別對個體單核苷酸多態性(SNP)進行檢測,并相互確認檢測結果。通過SAMtools軟件的“mpileup”程序進行序列校準,將覆蓋范圍在4~200的變異篩選出來進行后續分析。通過GATK軟件采用單體型檢測方法對每只綿羊的基因組變異進行檢測。采用過濾條件(MAF<0.05,丟失基因型>10%)過濾掉綿羊群體中具有較小等位基因頻率(MAF)的SNP,得到高可信度的SNP。
使用GATK軟件包對40個綿羊個體的基因組DNA序列進行插入或缺失(Indel)檢測,保留1-30 bp的Indel以備后續分析。
下載綿羊的SNP數據(https://www.ncbi.nlm.nih.gov/snp/),并與研究中檢測出的 SNP 進行比對驗證,確認上述過程檢測到的SNPs的準確性和可靠性。利用ANNOVAR軟件[12,13]注釋過濾和檢驗后的SNPs。
1.2 遺傳多樣性
使用 GATK(版本號:3.6-0-g89b7209)[11]軟件對4個綿羊品種基因組中的變異位點和基因型進行檢測,評估新疆細毛羊的遺傳多樣性。
1.2.1 雜合度(Heterozygosity,H)
采用mlRho程序[14]計算不同綿羊群體中每個個體的雜合度,作為評估不同綿羊品系群體間遺傳多樣性的指標之一。
1.2.2 連續純合子區域
使用PLINK軟件計算每個個體的連續性純合片段長度和數量,設置參數[15-17]:chr-set 26 -maf 0.05 -homozyg-window-snp 50 -homozyg-snp 50 -homo-zyg-kb 300 -homozyg-density 50 -homozyg-gap 1000 -homozyg-window-missing 5 -homozyg-window-threshold 0.05 -homozyg-window-het 03.基于 ROH 的物理長度,將 ROH 片段按照 <0.5 Mb、0.5~1 Mb、1~2 Mb、2~4 Mb、>4 Mb 進行分類統計,計算不同分類區間的ROH數目的比例。基于ROH計算基因組近交系數FROH,基因組近交系數的計算公式如下:
式中,∑LROH為常染色體上ROH片段的長度之和,Lgenome為常染色體的物理長度之和。
1.2.3 連鎖不平衡
使用Haploview軟件計算不同群體成對r2值,用以評估群體的連鎖不平衡程度,運用R軟件包繪制不同綿羊品種間連鎖不平衡衰減圖。
2 結果與分析
2.1 基因組DNA提取質量檢測
研究表明,DNA條帶單一且無拖尾,DNA樣品完整性較好、無嚴重降解。圖1
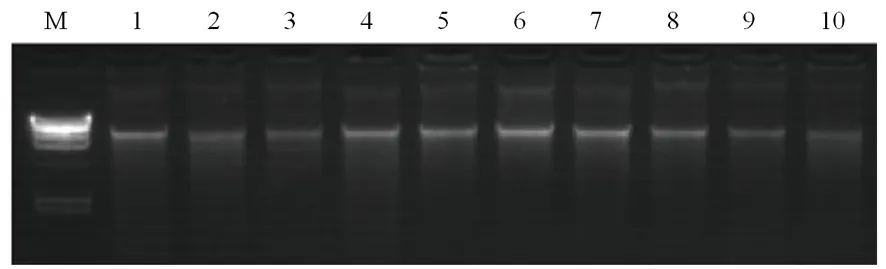
注:M 泳道為λ-HindⅢ DNA Marker,1-10泳道為DNA樣品
經試劑盒提取的基因組DNA OD260/OD280在1.8~2.0、基因組DNA濃度>50 ng/μL、基因組DNA總量>3 μg,基因組DNA的純度和濃度均符合測序要求。表1
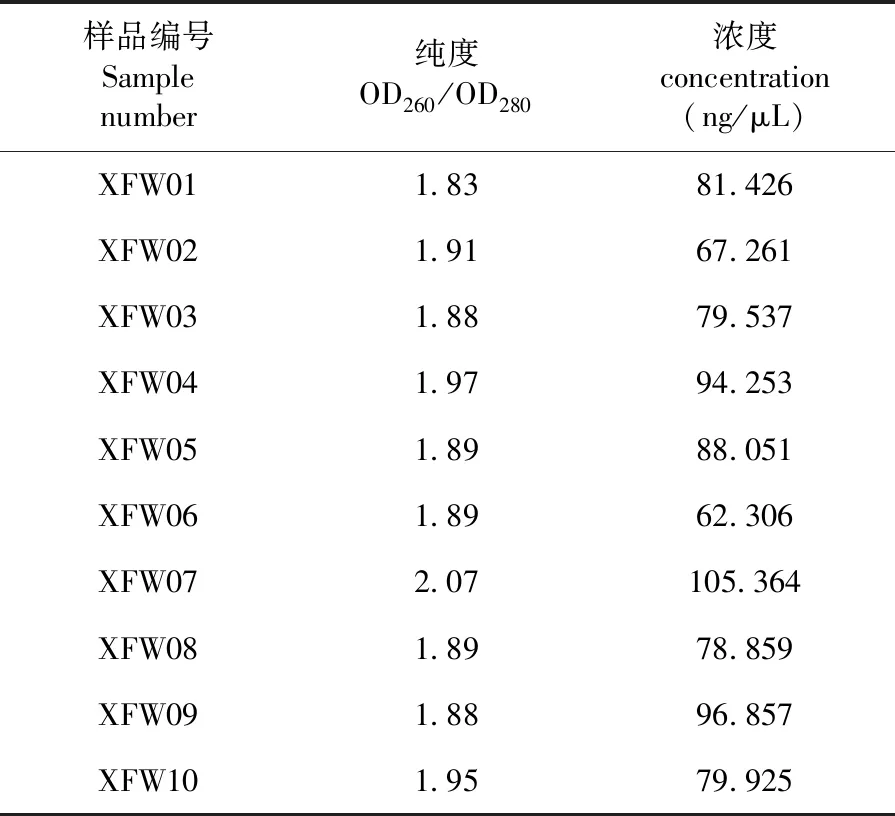
表1 新疆細毛羊DNA樣品濃度及純度檢測
2.2 全基因組測序數據量和質量統計
研究表明,得到的測序總深度高達76.84×,個體的平均測序深度為 7.68×,平均的比對率為96.38%,平均的基因組覆蓋度為97.88%,Q20(測序質量值≥20的堿基所占百分比)為96.97%,Q30(測序質量值≥30的堿基所占百分比)為91.93%。新疆細毛羊全基因組測序數據的比對率、全基因組覆蓋度、Q20、Q30等反映測序質量的數據值均較高,測序質量較好,測序數據能夠很好地反應個體的基因組信息。表2
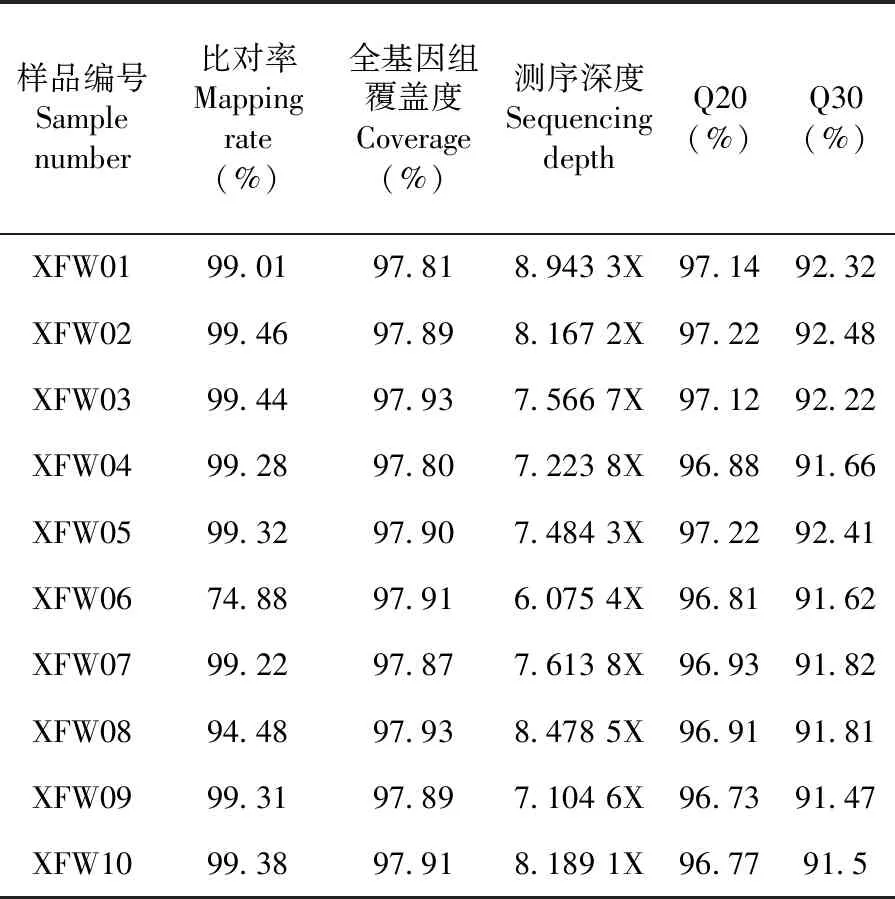
表2 全基因組測序數據質量統計
2.3 遺傳變異的鑒定、過濾和注釋
研究表明,最終獲得了97 647 435個高質量的常染色SNP位點和15 886 270個Indel;其中,34 744 752個SNP位點(35.58%)和5 588 596個Indel(35.18%)位于內含子區域。還發現了97 986個錯義變體和196 521個同義變體,以及1 394個缺失和981個插入導致的移碼突變。表3
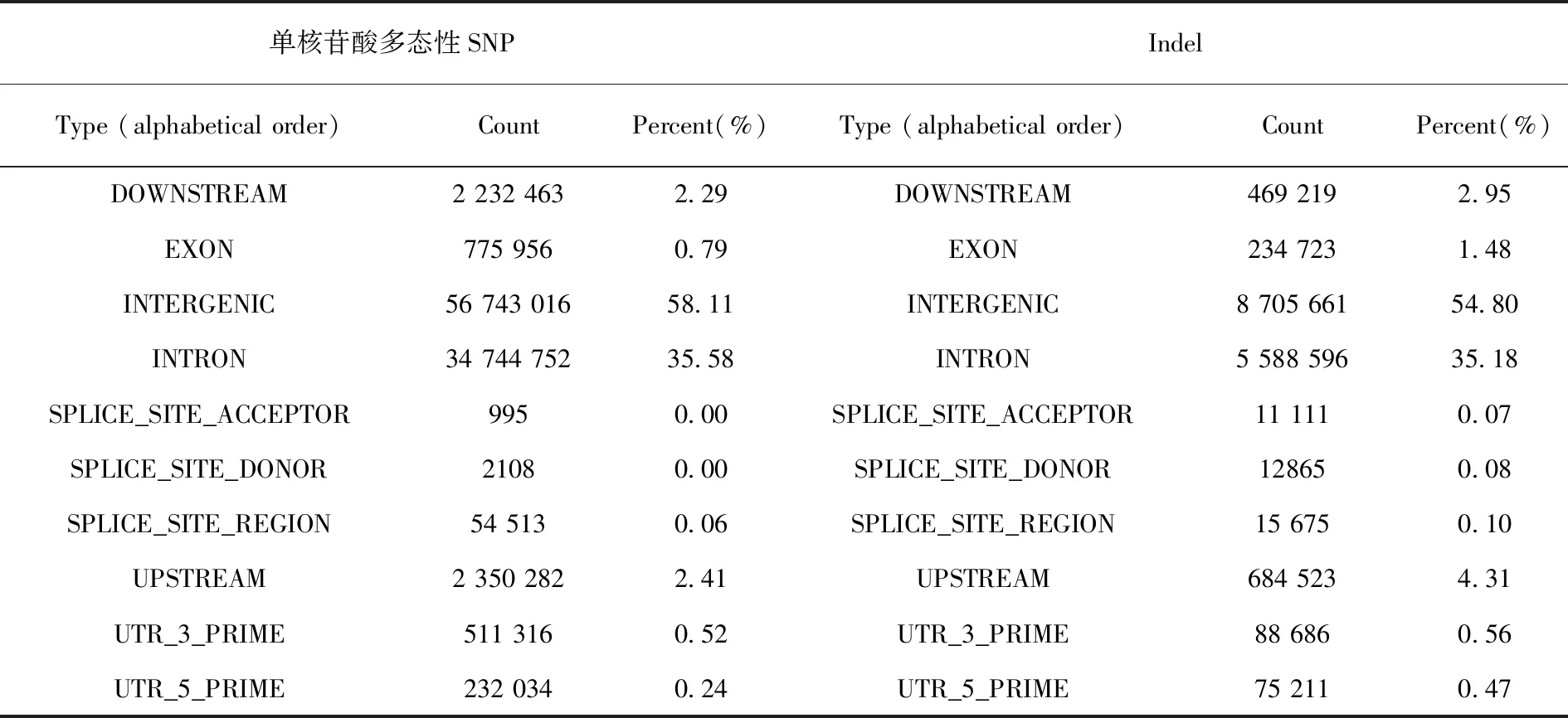
表3 新疆細毛羊全基因組重測序數據遺傳變異鑒定、過濾和注釋
2.4 4個綿羊群體的雜合度
研究表明,雜合度(Ho)和期望雜合度(He)的范圍分別為0.175~0.233和0.239~0.245。在所有群體中,期望雜合度略高于觀測雜合度,綿羊群體均有不同程度的近交或受到不同程度(人工或自然)選擇的影響。新疆細毛羊的平均觀測雜合度(Ho=0.196)極顯著低于阿勒泰羊(Ho=0.222)(P<0.001),低于巴音布魯克羊(Ho=0.201),略高于策勒黑羊(Ho=0.194)。圖2
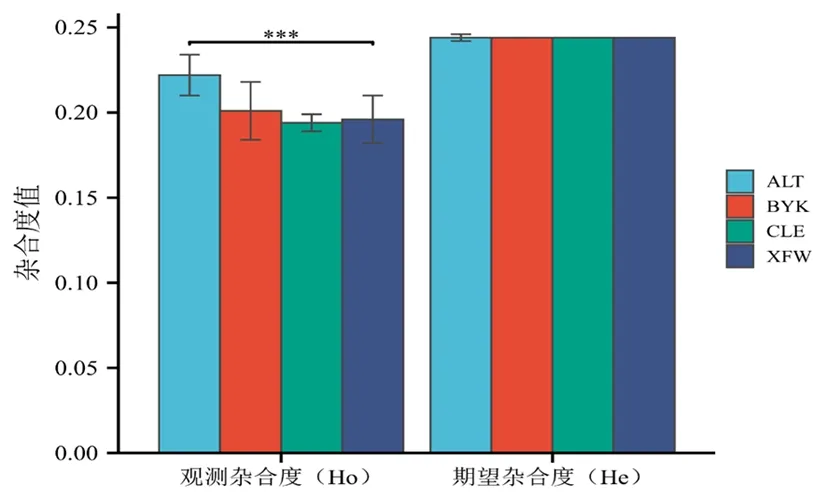
注:*表示不同綿羊群體間的雜合度具有顯著差異(P<0.05),**表示不同綿羊群體間的雜合度具有極顯著差異(P<0.01)
2.5 連續純合子區域差異
研究表明,4個綿羊群體間的ROH長度區間差異較大,在47.985~178.833 Mb,而新疆細毛羊、策勒黑羊、巴音布魯克羊、阿勒泰羊的平均ROH長度分別約為110.665、93.532、88.417和74.445 Mb,基因組近交系數分別約為0.042 3、0.035 8、0.033 8、0.028 5。表4
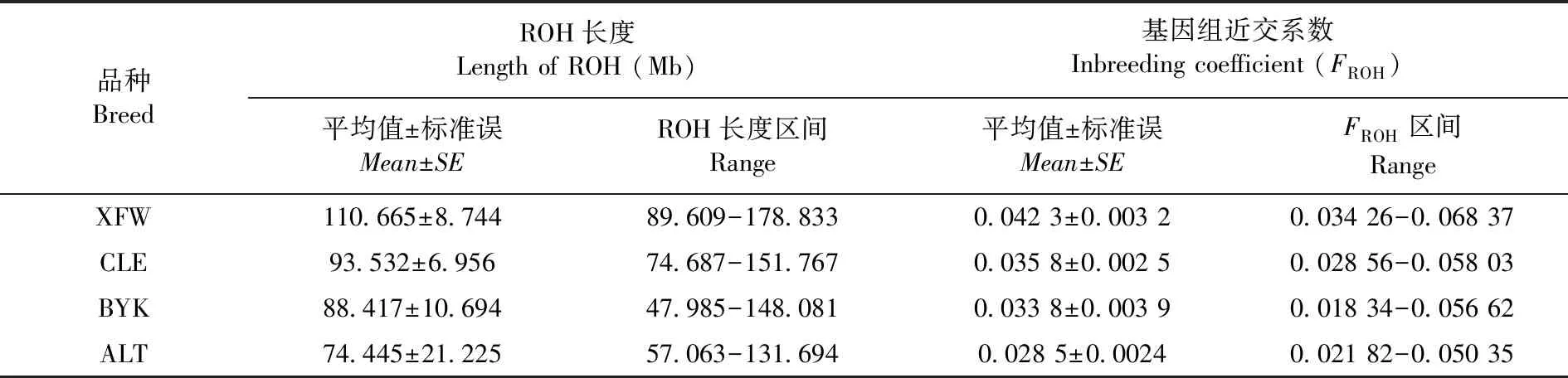
表4 4個綿羊群體的平均ROH長度和基因組近交系數
新疆細毛羊在<0.5 Mb范圍的ROH總長度顯著高于巴音布魯克羊、策勒黑羊和阿勒泰羊(P<0.05),新疆細毛羊遺傳多樣性水平低于巴音布魯克羊、策勒黑羊和阿勒泰羊。新疆細毛羊的平均ROH片段數量(215.8)顯著高于策勒黑羊(166.2)、巴音布魯克羊(152.9)和阿勒泰羊(150.2)(P<0.05),4個綿羊群體遺傳多樣性順序:阿勒泰羊> 巴音布魯克羊>策勒黑羊>新疆細毛羊。圖3,圖4
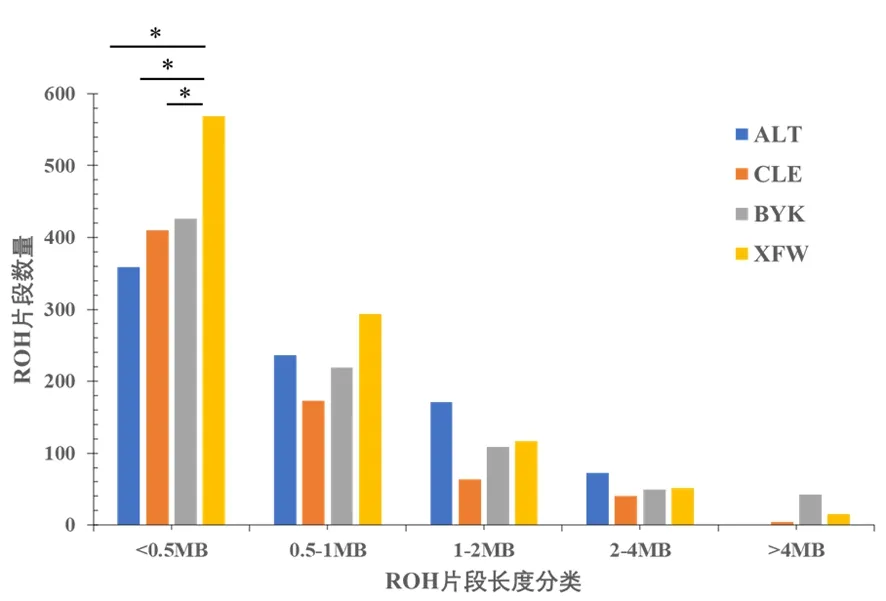
注:*表示不同綿羊群體間的ROH片段長度差異顯著(P<0.05)
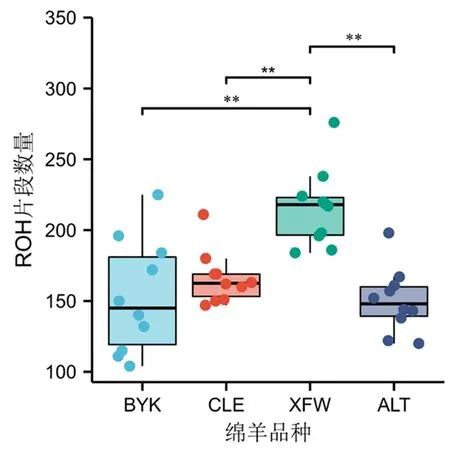
注:**表示不同綿羊群體間的平均ROH片段數量差異極顯著(P<0.01)
2.6 4個綿羊群體的連鎖不平衡性
研究表明,r2為0時,完全連鎖平衡,群體獨立遺傳;r2等于1時,表示完全連鎖不平衡。不同群體的綿羊整體連鎖不平衡程度都較低,其中,新疆細毛羊的連鎖不平衡程度相對最低,阿勒泰羊的連鎖程度相對最高的,其次是巴音布魯克羊和策勒黑羊,阿勒泰的遺傳多樣性最高,其次是巴音布魯克羊,新疆細毛羊的遺傳多樣性最低。圖5
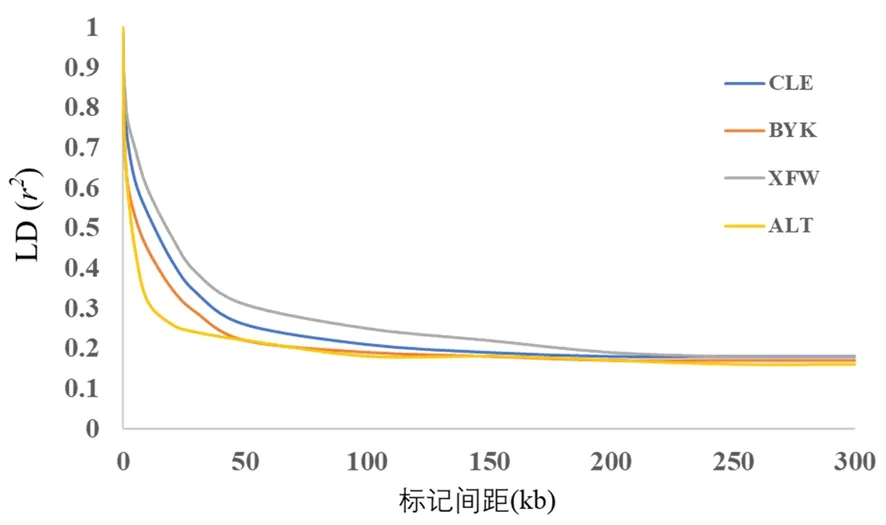
圖5 4個綿羊群體的連鎖不平衡變化
3 討 論
3.1 新疆細毛羊的全基因組重測序和遺傳變異檢測
單核苷酸多態性(Single Nucleotide Polymorphisms,SNP) 指在基因組上單個核苷酸的變異(包括置換、顛換、缺失和插入),形成遺傳標記,具有數量多,分布廣泛,多態性豐富,易于快速、規模化篩查,便于基因分型等特點,其作為新的遺傳標記對基因定位及相關疾病研究的意義亦非常重大。研究表明,部分基因的SNP位點與個體的發育性狀有關,可以將此類基因作為選育生長發育性狀的分子遺傳標記[18,19],有利于加速 SNP 分子標記技術在綿羊育種、遺傳分析中的應用[20]。研究采用全基因組重測序技術對10個新疆細毛羊個體進行全基因組重測序,通過遺傳變異鑒定和基因型信息整合,對變異位點質量值,測序深度,有效信息比例等一系列的質量過濾和控制條件,最終獲得了97 647 435個高質量的常染色SNP位點和15 886 270個Indel;其中,34 744 752個SNP位點(35.58%)和5 588 596個Indel(35.18%)位于內含子區域。此外,還發現了97 986個錯義變體和196 521個同義變體,以及1 394個缺失和981個插入導致的移碼突變。
3.2 新疆細毛羊的遺傳多樣性
3.2.1 雜合度
雜合度(Heterozygosity,H)是度量自然群體遺傳變異的首選指標,表示在一個群體中某位點為雜合子的概率。群體雜合度能反映群體的遺傳結構甚至是變化歷史,其值介于0到1。當某一群體的期望雜合度(He)高于其觀測雜合度(Ho)時,則群體被認為可能受到了選擇或者近交影響;當某一群體的期望雜合度(He)低于其觀測雜合度(Ho)時,則群體被認為可能引進了其他品種的血緣。雜合度越高意味著群體遺傳多樣性越豐富,反之,雜合度低群體遺傳多樣性低。新疆細毛羊的平均觀測雜合度(0.196)極顯著低于阿勒泰羊(0.222)(P<0.001),低于巴音布魯克羊(0.201),略高于策勒黑羊(0.194),新疆細毛羊的遺傳多樣性水平顯著低于阿勒泰羊,低于巴音布魯克羊,略高于策勒黑羊。
3.2.2 連續純合子區域分析
長純合片段(rus of homozygosity,ROH)是一類基因組中出現的連續不間斷的純合現象,表現為一段染色體區域缺乏雜合子[21]。在基因組某一段區域內,當一定數量一定密度的SNPs表現為純合時,可以判斷該區域存在ROH現象。遺傳漂變(Genetic Drift)、群體結構(Population Structure)、人工選擇(Artificial Selection)、連鎖不平衡(linkage Disequihbnum,LD)、近親交配( inbreeding)等都會影響ROH的產生,其中近親交配是影響ROH的首要因素[21,22]。不同的動物群體由于有著不同的群體大小、群體結構和交配體制,受到不同的自然或人工選擇方式、選擇強度影響,通過長期的歷史進化過程,都會在基因組上形成獨特的ROH模式,ROH可以反映不同動物群體所蘊含的獨特遺傳背景信息。ROH可以用于估計動物的近交情況[23,24]、推測近交的歷史[25,26]、鑒定受到選擇的基因[27,28]和有害突變[29-31]、評估遺傳多樣性和遺傳資源保護[23,32-34],以及優化動物育種規劃[29,35]。長的ROH片段反映最近世代發生過近交,而短的ROH說明較遠世代產生近交,因為世代數越短,ROH片段被重組打斷的可能性就越小。利用ROH計算基因組近交系數FROH可用于對物種或群體的近交情況進行評估,其計算方法為基因組中ROH片段的總長度占基因組總長度的比例。基因組近交系數FROH和系譜近交系數FPED呈中等程度相關或強相關,且基于ROH計算的基因組近交系數FROH最接近真實的近交系數[35-39]。
連續純合子區域分析結果顯示:新疆細毛羊、策勒黑羊、巴音布魯克羊、阿勒泰羊的平均ROH長度分別約為110.665、93.532、88.417 和74.445 Mb,基因組近交系數分別約為0.042 3、0.035 8、0.033 8、0.028 5。新疆細毛羊在<0.5 Mb范圍的ROH總長度顯著高于巴音布魯克羊、策勒黑羊和阿勒泰羊(P<0.05)。新疆細毛羊的平均ROH片段數量(215.8)顯著高于策勒黑羊(166.2)、巴音布魯克羊(152.9)和阿勒泰羊(150.2)(P<0.05),4個綿羊群體遺傳多樣性順序為:阿勒泰羊>巴音布魯克羊>策勒黑羊>新疆細毛羊。
3.2.3 連鎖不平衡分析
連鎖不平衡(Linkage Disequilibrium,LD)是指分屬兩個或兩個以上基因座位的等位基因同時出現在一條染色體上的幾率,高于隨機出現的頻率,呈現出一種相互關聯的現象[40,41]。重組率(Recombination),遺傳漂變(Genetic Drift),突變速率(Mutation Rate),自然選擇(Natural Selection),交配行為方式(Mating Behavior),遺傳連鎖方式(Genetic Linkage Mode)等許多因素都會對連鎖不平衡產生影響。不同物種或群體所經歷的特殊歷史事件及繁殖行為等特征可以通過連鎖不平衡情況進行反映。馴化選擇,會導致群體遺傳多樣性下降,位點間的相關性(連鎖程度)加強。通常馴化程度越高,選擇強度越大的群體,LD衰減速度是最慢的。連鎖不平衡分析結果表明:阿勒泰羊的連鎖不平衡程度相對最低,新疆細毛羊的連鎖程度相對最高的,其次是策勒黑羊和巴音布魯克羊,該結果與雜合度和連續純合子區域分析的結果基本一致,阿勒泰的遺傳多樣性最高,其次是巴音布魯克羊,新疆細毛羊的遺傳多樣性最低。
4 結 論
新疆細毛羊的平均觀測雜合度(Ho=0.196)極顯著低于阿勒泰羊(Ho=0.222)(P<0.001),低于巴音布魯克羊(Ho=0.201),略高于策勒黑羊(Ho=0.194);新疆細毛羊在<0.5Mb范圍的ROH總長度顯著高于巴音布魯克羊、策勒黑羊和阿勒泰羊(P<0.05),其平均ROH片段數量(215.8)顯著高于策勒黑羊(166.2)、巴音布魯克羊(152.9)和阿勒泰羊(150.2)(P<0.05);在4個綿羊群體中,新疆細毛羊的連鎖不平衡程度相對最低。新疆細毛羊的遺傳多樣性水平相對低于阿勒泰羊、巴音布魯克羊和策勒黑羊。
猜你喜歡
國畫家(2022年2期)2022-04-13 09:07:46
四川文學(2021年4期)2021-07-22 07:11:54
新作文·高中版(2021年4期)2021-07-14 11:55:06
環球人物(2021年8期)2021-05-06 08:00:43
音樂天地(音樂創作版)(2020年12期)2021-01-11 01:13:40
音樂天地(音樂創作版)(2020年7期)2020-09-11 06:17:18
華人時刊(2018年15期)2018-11-10 03:25:32
人民中國(日文版)(2015年10期)2015-04-16 03:53:48
絲綢之路(2014年9期)2015-01-22 04:24:46
新疆人文地理(2009年7期)2009-09-29 09:56:14
