肝豆狀核變性發病機制、臨床表型-基因型關系及藥物治療研究進展
2023-07-05 02:57:14瑪力帕提艾爾肯江凱迪日亞庫爾班徐玲孫曉風
臨床肝膽病雜志 2023年6期
瑪力帕提·艾爾肯江, 凱迪日亞·庫爾班, 徐玲, 孫曉風
新疆醫科大學第一附屬醫院感染科, 烏魯木齊 830000
肝豆狀核變性又稱 Wilson 病(WD),是一種常染色體隱性遺傳病,由位于13號染色體上的ATP7B基因突變引起銅(Cu)排泄減少。過量的Cu會導致多種臨床表現,包括神經系統癥狀、急性或慢性肝衰竭和/或精神癥狀。1932年,Cheng[1]首次報道了中國的兩例WD病例,WD的流行程度因地理區域而異,亞洲國家的發病率比西方國家高[2]。WD的常規診斷是根據其臨床癥狀和血銅藍蛋白(CP)、24 h 尿Cu、角膜 K-F環等生化指標作為篩查指標,Leipzig評分可以提高診斷效能。但在大多數受影響的患者中,WD表現為肝功能障礙或不明原因的CP水平降低,加之WD的臨床表現多樣化,早期診斷困難。此時,應行肝組織病理學檢查和 ATP7B 基因檢測聯合分析[3]。而基因診斷有著重要的臨床意義,并在我國逐漸成為一種常規檢測[4]。經過規范化治療,患者預后良好,與普通人無差異。因此,WD的早期診斷和規范治療至關重要,以防止疾病惡化[5]。目前絕大多數患者需要終生服藥來控制Cu在其體內的吸收和儲存。
1 發病機制
1.1 人體內的Cu穩態 人們每天從飲食中攝入1.5~5 mg的Cu,其中50%~ 60%未吸收的Cu隨糞便排出;25%~40%從十二指腸吸收,由腸細胞儲存。在腸道池中,Cu與循環中的白蛋白結合,75%運輸到肝臟進行調節和排泄,剩下的25%進入血液循環。在肝臟中,20%(2.5 mg/d)的Cu通過膽汁排泄且其中很大一部分(約80%)再次被腸黏膜重新吸收,少量(20%)隨糞便排出。80%的Cu被轉運到外周,與CP結合。因此,Cu每天的糞便排泄是未吸收的攝入Cu和少量排出的內源性Cu的組合,為1.5~4 mg/d。
Cu是許多酶的輔助因子,是正常新陳代謝所必需的。如參與呼吸作用[如細胞色素C氧化酶(COX)]、神經內分泌肽(如肽基-α-單加氧酶)的激活、色素沉淀(如酪氨酸酶)、兒茶酚胺的合成與清除(如多巴胺-β-單加氧酶)、自由基防御[如超氧化物歧化酶(SOD)1和SOD3]以及許多其他的細胞反應過程[6]。Cu的攝取是通過銅轉運蛋白1(CRT1)發生在肝細胞基底外側。Cu伴侶將Cu運送到特定的細胞內靶點,例如:SOD的Cu伴侶將Cu運送到SOD;線粒體中的一組伴侶蛋白(Cos11、Cos17、SCO1和SCO2),將Cu傳遞給COX;在反式高爾基網絡(TGN)中,Cu轉運蛋白(ATOX1)將Cu轉運至Cu轉運ATP酶1(ATP7A) 和跨膜Cu轉運ATP酶2 (ATP7B)。ATP7B有助于將Cu轉運到TGN和Cu的膽道排泄。在肝細胞內,ATP7B在TGN或細胞質囊泡中發揮兩種重要功能。在TGN中,ATP7B通過將6個Cu分子運輸到原血漿CP中,激活功能性CP。在細胞質中,當細胞內Cu離子水平升高時,ATP7B 將多余的Cu釋放到囊泡中,并通過胞外排出,穿過根尖管膜進入膽汁。在這一過程中,ATP7B借助 ATP 水解釋放的能量磷酸化,隨后脫磷酸化釋放Cu跨膜所需的能量[7]。因此,ATP7B 依賴性膽汁Cu排泄是維持Cu代謝平衡的主要方式(圖1)。
1.2 Cu毒性相關機制 WD是由編碼P型ATP酶的ATP7B基因缺陷引起的。該蛋白的作用是將Cu裝載到高爾基體中原血漿CP上,在肝細胞中成為功能性CP。CP經膽道排泄,作為清除體內多余Cu的主要途徑。在WD中,ATP7B蛋白的功能障礙導致無法將Cu裝載到高爾基體中的原血漿CP上,從而不能適當地將Cu排泄到膽汁中。結果,Cu在肝細胞中積聚,隨后釋放到血液中。血液中存在兩種Cu池:與CP結合(85%~95%)和非CP結合或游離銅(5%~15%),后者與白蛋白松散結合,可被細胞攝取或參與代謝[8]。原血漿CP不與Cu結合,在血流中迅速降解,導致WD血清CP水平低。正常情況下,由于大多數血清Cu與CP結合,而在WD中CP水平較低,因此該病血清總Cu水平較低,游離Cu水平升高。
Cu的細胞毒性主要與其氧化還原特性有關。未結合的“游離”Cu離子與活性氧將通過芬頓樣(Fenton-like)反應導致對蛋白質、核酸和脂質高度有害的羥基自由基的出現。在亞細胞水平上,線粒體是Cu誘導毒性最敏感的靶點,線粒體利用細胞內的Cu進行呼吸,是細胞內Cu平衡的關鍵調節器。在細胞中,Cu與蛋白質或小分子配體嚴格結合并作為線粒體COX或SOD1的輔助因子,COX或SOD1分別處理Cu介導下的氧化還原或歧化反應[9]。故當體內Cu離子超負荷時,Cu的氧化還原活性不受控制,導致線粒體膜的結構、物理和生化完整性遭到破壞。然而對于WD,找到是否存在特定的線粒體Cu排泄途徑來平衡線粒體Cu超載,在未來治療中是非常可期的。有研究[10-11]認為,游離Cu的毒性作用也與Cu誘導的凋亡抑制劑功能障礙和caspase-3失控有關。
1.3 ATP7B分子結構和功能 ATP7B蛋白屬P型ATP酶超家族,由位于第13號染色體長臂(13q14.3~q21.1),全長約85 kb的ATP7B基因編碼。P型ATP酶超家族有11類。IB類P型ATP酶負責運輸Cu2+和其他重金屬離子穿過生物膜[12]。ATP7B有8個跨膜螺旋。它的總體結構包括:一個大的N端結構域,其包含6個子結構域,每個子結構域都有一個金屬結合位點(MBD);ATP結合結構域由核苷酸結合域和磷酸化域組成;一個執行器結構域(A-結構域);一個長長的C端尾。ATP7B與Cu在N端結構域結合,MBD在從Cu伴侶ATOX1接受Cu的過程中發揮核心作用。研究表明MBD對ATP7B活性的影響不等,MBD5和MBD6對ATP7B的催化活化作用強于MBD1~4[13],并以ATP作為能量源,將其跨細胞膜運輸。
綜上可知,ATP7B維持細胞內Cu穩態,一旦其功能和結構受損,CP水平降低,肝臟出現Cu超載及發揮其Cu毒性,導致WD的各種臨床表現。
2 ATP7B基因突變類型與臨床表型關系
WD具有復雜的臨床表型,現已有超過900多個ATP7B基因突變被發現。其中380個已經證實與疾病的發病機制有關[14]。大多數是錯義突變,這些突變對ATP7B 蛋白結構(例如,ATP7B完整性的喪失、錯誤折疊、蛋白間相互作用受損)和功能(磷酸化、Cu離子運輸異常、ATP結合親和力下降和細胞內運輸異常)產生了不同程度的影響[15]。
在歐洲和亞洲人群中,最常見的突變分別是p.His1069Gln和p.Arg778Leu,其中復合雜合子比純合子更為常見。在Das等[16]對純合條件下p.His1069Gln和p.Arg778Leu進行的一項基因型-表型Meta 分析中顯示,p.Arg778Leu患者的發病年齡早,大多表現為肝臟癥狀和高尿Cu。p.His1069Gln患者主要表現為遲發性神經系統癥狀 (平均發病年齡計算為20歲)。在張天鶴等[17]納入的70例兒童肝豆狀核變性的臨床特點及基因分析的一項研究中顯示,p.Arg778Leu突變的患者,血清CP水平較其他突變更低,多數表現為肝臟癥狀,差異具有統計學意義(P<0.05)。一項納入了55例WD患者的研究[18]中顯示p.Arg778Leu突變可能在更早的年齡導致更嚴重的肝損害;一項對一個大型黎巴嫩近親家庭76例成員的研究[19]表明,c.2299insC與肝臟疾病之間存在關聯,p.Ala1003Thr突變與神經疾病之間存在關聯。
而Zhang等[20]發表的一項共納入1 366例中國WD患者的評估基因型-表型相關性的研究鑒定出了294例潛在致病性ATP7B 變異,包括116例新變異,其中48個可歸為 “致病變異。變異頻率依次為c.2333G>T (p.Arg778Leu)、c.2975C>T (p.Pro992Leu)、c.2621C>T (p.Ala874Val),并發現這些變異與肝臟或神經表現無關,而p.Ala874Val是構音障礙相關的獨立因素。此外,這項研究強調了WD患者在發病年齡和 ATP7B變異方面的差異,早發型患者以PTV(蛋白質截斷變異)、p.Arg778Leu、p.Pro992Leu為主,晚發型患者以p.Ala874Val為主,且p.Arg778Leu/p.Ala874Val基因型患者的發病年齡高于p.Arg778Leu/p.Arg778Leu或p.Arg778Leu/p.Pro992Leu基因型患者。在一項來自印度不同地方的針對58例WD患者進行的研究[21]顯示,最常見的變異為 c.813C>A (p.Cys271Ter)并無法表明基因變異-臨床表型的關系,認為額外的因素或基因可能在WD 的病因中起作用。最近,劉攀等[22]對75 例WD患兒進行了回顧性分析,結果顯示該研究中最常見的突變類型為p.Arg778Leu、p.Ala874Val、p.Pro992Leu,并鑒定出了6個未報道的ATP7B新變異,且發現基因型-表型無明顯相關性(表1)。
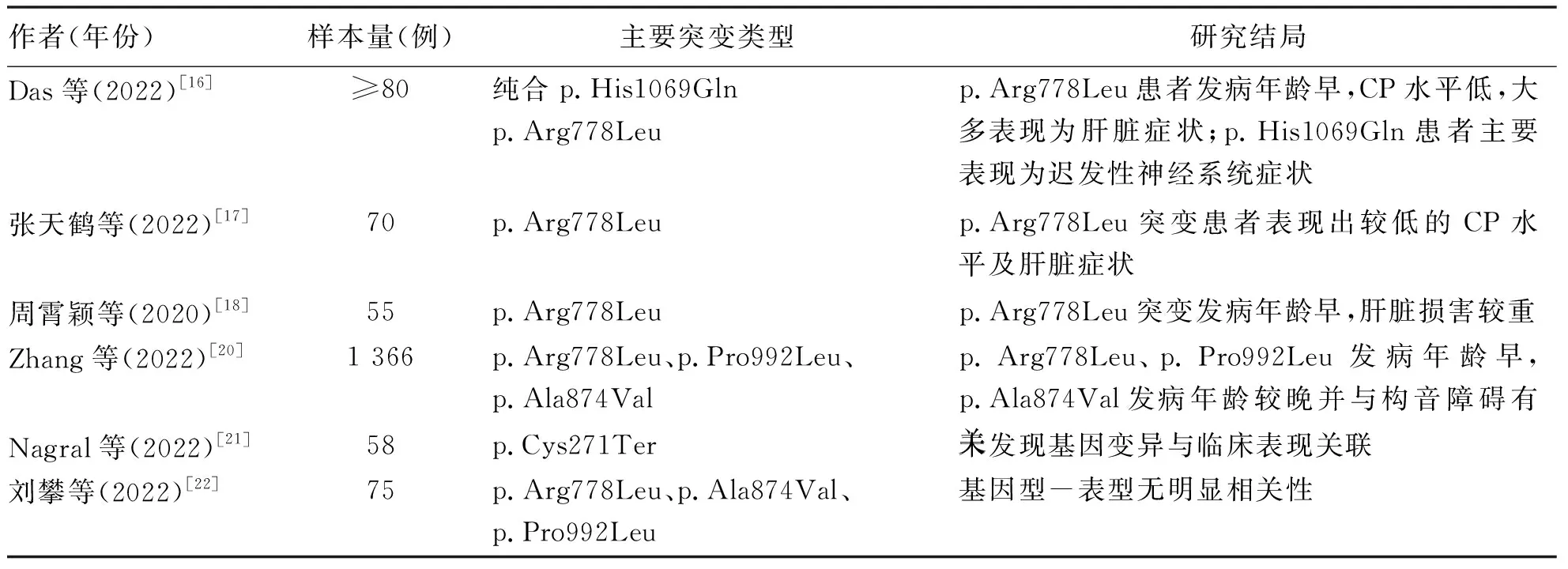
表1 ATP7B基因突變類型與臨床表型關系Table 1 Relationship between ATP7B gene mutation type and clinical phenotype
總體而言,試圖進行基因型-表型相關性研究的結果尚未得出明確的結論,主要原因是WD患者的表型特征較差,診斷較晚。遺傳和環境因素也可能相互作用影響復雜的疾病表型;然而,還需要進一步的大型研究來提供具體關聯的結論性證據[6]并需與其他Cu代謝指標異常的遺傳性疾病正確鑒別,具體見表2。除了ATP7B異常突變外,Cu代謝結構域蛋白1、ATOX1、四氫葉酸亞甲基還原酶、凋亡抑制基因蛋白、人含 Patatin 樣磷脂酶域蛋白 3、二價金屬轉運蛋白1等異常亦可與改變WD的表型有關,但仍需進一步證明診斷或預測價值[2]。
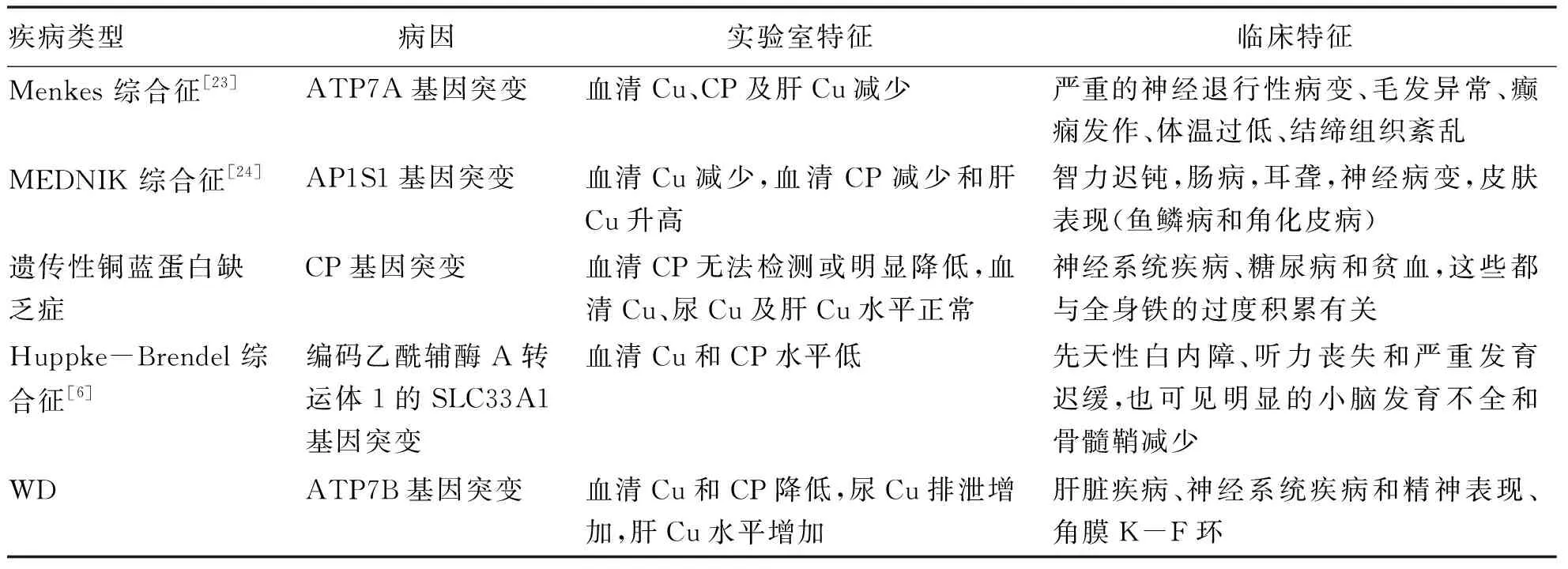
表2 WD與其他Cu代謝指標異常遺傳性疾病的鑒別Table 2 Differential diagnosis of WD and other genetic diseases with abnormal copper metabolism indexes
3 藥物治療進展
治療的總目標是通過平衡Cu的攝入和排出來建立正常的Cu穩態。總結治療方法:第一,如果早期診斷(在嚴重的肝臟或神經損傷之前),并且終生堅持治療,WD可以成功地通過藥物來控制病情進展;第二,應根據可能發生的不良事件進行藥物的選擇;第三,對于正在接受藥物治療的WD患者,每年至少應進行兩次監測,以確定治療在臨床改善和生化變化方面的有效性,從而評估治療依從性并發現任何治療誘導的不良事件 (包括血清Cu、24 h 尿Cu、血清CP、肝功能、神經和精神狀態以及疾病對其他器官的任何影響);第四,如果需要,對肝臟、神經或精神癥狀進行對癥治療,并隨著時間的推移進行重新評估[6]。目前的治療方法包括藥物治療、肝臟移植、基因治療及細胞療法。下面對藥物治療方面的進展進行綜述。
目前治療WD的藥物包括傳統螯合劑(青霉胺、曲恩汀、二羥丙磺酸鈉及二羥丁二酸),增加尿Cu排泄;鋅鹽及新型螯合劑(四硫代鉬酸銨、雙膽堿四硫鉬酸鹽),減少Cu的消化道吸收。目前指南建議使用螯合劑(青霉胺和曲恩汀)作為有癥狀WD患者的首選治療[14]。青霉胺是國內治療WD患者的一線藥物。由于其成本低,購買渠道方便,仍然是最常用的驅Cu劑。青霉胺在肝功能障礙患者中,通常在治療的前2~6個月內出現臨床改善,可在治療開始1年后實現完全緩解。在神經障礙患者中,WD癥狀和體征消失較慢,緩解可能需要長達3年。兒童使用青霉胺的劑量是20 mg·kg-1·d-1,最大劑量為 750 ~ 1 000 mg/d。成人初始劑量為 125 ~ 250 mg/d,每 4~7 d 增加250 mg/d,維持劑量為750~1 500 mg,分2~3次劑量。食物會減少50%的吸收,所以在用藥前1 h和用藥后2 h都不能給食物。青霉胺作為吡哆辛拮抗劑可增加尿液中的維生素排泄。因此,建議每天補充25~50 mg維生素B6,以預防因吡哆辛缺乏而可能發生的貧血和/或周圍神經炎癥[25]。青霉胺有諸多不良反應:包括發燒、皮疹、骨髓抑制、淋巴結病、蛋白尿、腎毒性、腎病綜合征、視神經炎、視網膜炎、紅斑狼瘡、重癥肌無力、多發性肌炎等[26],Emre Parlar 等[27]報道了1例使用青霉胺治療WD并在治療過程中出現彌散性血管內凝血(disseminated intravascular coagulation, DIC)的患者,需要注意的是,在WD患者出現出血性并發癥的情況下,在排除其他常見原因后,青霉胺可以引起血小板損傷,通過激活血管內凝血導致內皮損傷和DIC。此外,最令人擔憂的情況之一是在治療開始時出現嚴重和不可逆的神經系統惡化,它是由于在螯合劑治療后Cu從肝臟釋放而引起氧化性腦損傷。這可能發生在 10%~50%既往有神經系統癥狀的患者[28]。在這些情況下,應該停用青霉胺,用其他藥物替代。
曲恩汀(三乙烯四胺)是WD的替代螯合劑。它是精胺和腐胺的衍生物,以1∶1的比例與Cu結合形成穩定的絡合物[29]。具有與青霉胺相似的作用機制和治療效果。該藥不穩定,需在 2~8 ℃條件下保存,價格昂貴,目前國內尚未生產。曲恩汀的兩種配方二鹽酸曲恩汀和四鹽酸曲恩汀,已經在部分歐美國家被批準使用,這兩種產品都可以在室溫下儲存。曲恩汀治療劑量為1 g/d, 分2~4次 , 維持劑量為 0.75~1 g/d。兒童用量按 20 mg·kg-1·d-1計算 。服藥次數越多,依從性越差。Ala等[30]的一項前瞻性研究,共納入了8例年齡在22~71歲的WD患者,所有患者接受每日單劑量曲恩汀(15 mg/kg),并持續監測至第12個月。結果顯示,所有患者臨床癥狀均良好,沒有發現新的神經體征,沒有停止治療或退出研究的情況。故認為,應進一步研究每日一次的曲恩汀作為WD的維持治療。曲恩汀的潛在副作用包括全血細胞減少、出血性胃炎、味覺喪失、系統性紅斑狼瘡和神經系統惡化[25]。 曲恩汀也可能導致神經性WD的惡化,因此,應該從低劑量開始,并像青霉胺一樣緩慢增加。曲恩汀不能和鐵一起服用,因為它會形成有毒復合物。口服青霉胺的驅Cu效果遠高于曲恩汀,一項對接受曲恩汀和青霉胺的患者的大型回顧性研究[31]表明,這兩種藥物的療效相似,但青霉胺組的副作用較大。Weiss等[32]的研究表明,對青霉胺停藥后的WD患者,鹽酸曲恩汀是耐受性良好的治療方法。曲恩汀是一種有效的替代Cu螯合劑,與青霉胺治療相比,曲恩汀的不良反應相對較少,因而被推薦用于治療對青霉胺不耐受的WD患者[33]。
二巰基丙磺酸鈉(DMPS)及二羥基丁二酸是在我國開創性用于治療WD的廣譜金屬螯合劑。其作用機制為分子中的2個羥基與Cu離子結合,形成毒性較低的硫醇化合物后經腎小球濾過,最終通過尿液排出體內。前者驅Cu作用高于青霉胺,主要適用于對青霉胺過敏或出現嚴重的錐體外系癥狀的患者。最新指南建議DMPS成人劑量為500~750 mg,可分次靜脈推注(3 次/d)或 滴 注(1 次/d),5 d為1 個療程,兒童劑量為 10~20 mg·kg-1·d-1。后者驅Cu作用較青霉胺弱,主要適用于輕-中度肝臟損害、神經精神癥狀及對青霉胺不耐受的 WD 患者,成人推薦劑量為 750~1 000 mg/d,2 次/d;兒童推薦劑量為10~20 mg·kg-1·d-1,2 次/d。兩種藥物均可與鋅劑聯合使用,也可與青霉胺、鋅劑交替使用。主要不良反應包括胃腸道反應、皮疹、發熱、瘙癢等過敏性癥狀。需注意的是,由于DMPS不能通過血腦屏障,可能導致少數WD患者神經表現惡化[34]。
鋅試劑對WD的治療有明確的療效,副作用較少,最常見的副作用是刺激胃腸道。鋅的作用機制包括刺激腸黏膜上皮內金屬硫氨酸的合成(一種內源性蛋白質金屬螯合劑)。這種蛋白質優先結合腸細胞中的Cu,并抑制其滲透到門靜脈循環。另一種作用是誘導肝臟金屬硫蛋白結合Cu,防止其毒性損傷肝細胞。不同于青霉胺和曲恩汀,鋅的作用是通過增加糞Cu的排泄。它的主要缺點是通常需要4~6個月才能發揮作用。因此,對于病情嚴重的WD患者,它并不是首選,主要是作為維持治療。但對于癥狀前患者、孕婦和肝移植術后患者,一直作為一線治療。鋅鹽成人治療劑量為50 mg/d,3次/d;5 歲以下兒童治療劑量為 25 mg/d,2次/d;5~15 歲兒童治療劑量為25 mg/d,3次/d。 餐前或餐后 1 h 空腹服用。Avan等[35]認為,由于鋅毒性水平很低,不良反應小,可被推薦作為神經系統癥狀的一線治療方法。醋酸鋅是唯一被美國食品藥品監督管理局和歐洲藥品質量管理局批準用于治療WD的鋅鹽。由于副作用和價格較高,部分患者更傾向于使用葡萄糖酸鋅或硫酸鋅[14]。Munk等[36]的一項關于口服鋅方案對人肝臟Cu含量影響的研究中認為,WD標準治療醋酸鋅方案(50 mg×3)與葡萄糖酸鋅方案(50 mg×3)對肝臟Cu含量的降低程度相似。
目前尚不清楚鋅和螯合劑聯合治療是否比單一治療更有效,在Chen等[37]對涉及1 056例患者接受螯合劑與鋅聯合治療WD的一項Meta分析中指出,與肝臟癥狀為主的患者相比,神經系統表現的患者更有可能從聯合治療中獲益。因此,對于接受聯合治療的WD患者,臨床醫生應密切監測生化指標結果和臨床療程,特別是對肝臟表現的反應。
四硫鉬酸鹽是一種強力的除Cu劑,Ⅲ期臨床試驗結果[38]證實,四硫鉬酸鹽本身不會使神經功能惡化,因此特別適用于神經受累的WD 患者。但四硫鉬酸鈉在伴有肝病的WD患者中的應用還存在一定的局限性,加上藥物本身的不穩定性,并且伴有骨髓抑制、貧血和轉氨酶升高等相關副作用,目前未被批準上市。雙膽堿四硫鉬酸銨(TTM)是一種新型藥物, 其作用機制包括形成更穩定的Cu-TTM-白蛋白復合物,抑制肝臟和神經元Cu的攝取及促進Cu膽汁排出。此外,雙膽堿TTM可以通過血腦屏障并進一步進入神經元細胞。由于傳統螯合劑不能通過血腦屏障,因此,雙膽堿TTM可能成為一種新的治療策略,尤其對神經癥狀為主的WD患者有用[39]。雙膽堿四硫鉬酸銨臨床Ⅲ期試驗目前正在國外進行其長期療效和安全性的評估。
甲烷氧化菌素是一種新型治療WD的方法,目前正在進行研究,并在大鼠模型中取得了積極的結果。它可以從線粒體中去除Cu,避免細胞毒性和急性肝衰竭[40]。
4 總結與展望
WD患者的主要表現為肝臟、神經系統或精神癥狀。通常,神經系統癥狀出現較晚。肝臟癥狀主要出現在生命的早期,并可發展為嚴重的黃疸和肝硬化。ATP7B 是目前已知的導致WD的唯一基因,基因型-表型的相關性已經被廣泛研究,但直接原因仍未明確。未來需要更多精確、高質量、大規模的臨床驗證研究。進一步了解WD的發病機制,有助于明確WD 患者的早期診斷和制定相應的個體化治療方案,改善預后并避免誤診。
藥物治療仍然是控制WD的首要措施,但由于多數藥物存在較大的副作用,因此,今后需要投入更多的前瞻性臨床研究來尋找低毒性、高驅Cu作用的藥物。此外,對癥治療和康復培訓也是必不可少的。癥狀的明顯改善提高了生活質量,增加了患者對治療的信心和依從性。鼓勵和幫助患者以樂觀的態度參加適度的活動,恢復社會功能。目前基于基因檢測的新興醫學治療包括干細胞移植和基因療法的發展對今后WD的治療很有希望。
利益沖突聲明:本文不存在任何利益沖突。
作者貢獻聲明:瑪力帕提·艾爾肯江負責查閱文獻及撰寫論文;凱迪日亞·庫爾班、徐玲負責修改文章;孫曉風負責擬定寫作思路,指導撰寫文章并最后定稿。
猜你喜歡
課堂內外·初中版(科學少年)(2023年10期)2023-12-10 00:43:06
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
全科護理(2022年10期)2022-12-26 21:19:15
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
保健醫苑(2022年1期)2022-08-30 08:39:40
鄉村科技(2021年33期)2021-03-16 02:26:54
國際放射醫學核醫學雜志(2021年10期)2021-02-28 08:41:58
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
獸醫導刊(2016年6期)2016-05-17 03:50:35
