微波對大豆蛋白氧化聚集體結構及功能特性的影響
2023-07-14 14:28:24江連洲王一暢馬依彤楊宗瑞郭增旺
農業工程學報 2023年9期
關鍵詞:大豆
江連洲 ,王一暢 ,馬依彤 ,劉 軍 ,楊宗瑞 ,郭增旺 ※
(1. 東北農業大學食品學院,哈爾濱 150030;2. 山東禹王生態食業有限公司,德州 253000;3. 克東禹王大豆蛋白食品有限公司,齊齊哈爾 161000)
0 引言
大豆分離蛋白(soy protein isolate,SPI)是一種由脫脂大豆粕為原料生產的蛋白質質量分數達90%以上的食品原料[1],因其具有優良的功能性質和生理活性等特點而被廣泛應用于食品工業中[2]。目前,中國大豆分離蛋白年生產能力已達70 萬t,總產值突破百億,居世界第一。當儲存和運輸時間過長時,大豆分離蛋白的氧化改變了理化性質,導致其加工特性劣變,并最終影響其在食品加工品質中的應用[3]。脂質過氧化會產生各種活性氧成分物質(reactive oxygen species,ROS),它們已被證實是導致蛋白質氧化的重要因素[4]。一些研究表明,氧化修飾可引起蛋白質多肽骨架和氨基酸殘基側鏈的一系列變化,促進二硫鍵的形成并加強蛋白質的聚集,暴露后分子內的基團重組形成低聚物,在疏水性和靜電吸引作用下進一步形成大分子聚集體,導致其溶解性和結構柔性降低,進而使功能性質劣化[5]。且氧化程度越高,蛋白質變性越劇烈,其功能特性的變化越明顯。因此,調控蛋白質分子的聚集程度是改善蛋白質氧化聚集體功能活性下降的重要解決方式。
目前,許多研究工作都集中在使用物理方法通過加熱、機械作用等改變蛋白質的結構和聚集程度,實現對蛋白質聚集體的調控。文獻[6]表明,高壓均質使蛋白質解聚從而提高了蠶豆蛋白質的溶解度。文獻[7]表明,超聲處理通過調控蛋白質聚集度的不同導致其不同的理化和功能特性。但是,由于其巨大功率消耗和較低的生產能力,高壓均質化和超聲波工藝很難在食品工業中廣泛使用[8]。微波作為一種物理加工技術,由于其綠色、高效、簡單的操作方式,更適合在食品工業中應用[9]。微波改性的機制主要是因為偶極和離子振動生熱,食品物料的帶電性和極性基團在微波場下的響應不同,會產生振動,旋轉,最終表現為微波的靶向加熱,進而改變物料的理化和功能性質。現有研究表明微波處理能提高蛋白的乳化性、起泡性和交聯特性等功能特性,同時微波加熱的特殊效應還能夠改變蛋白的構象。一方面是因為微波交變電場下蛋白質氨基酸殘基上的帶電基團發生波動,導致蛋白質分子間的電場和靜電相互作用重新分布[9]。另一方面蛋白質帶電基團在微波場下的特殊響應也會對蛋白質的功能特性產生影響。文獻[10]發現微波處理有助于形成緊湊的網絡結構,蛋白質結構剛性和柔性區域的位置被改變。文獻[11]表明微波誘導了蛋白質極化和非極性基團的暴露,蛋白質柔性區域增加,從而使起泡性得到改善。文獻[12]研究表明大米蛋白受到微波處理后其內部的游離巰基減少,生成了新的二硫鍵,增強了蛋白質凝膠網絡的強度,進而改變大米的加工特性。這些研究均表明微波處理工藝能通過改變蛋白的聚集結構和分子構象來改變其功能特性。但是目前,關于微波對氧化聚集體的結構和功能活性作用間的關系,尤其是微波對大豆蛋白氧化聚集體的影響鮮有報道。
偶氮二異丁脒鹽酸鹽(2,2′-azobis (2-amidinopropane)dihydrochloride,AAPH)是一種具有良好可控性、穩定性、重復性和適用性的自由基引發劑,因其不含腈基,分解產物無毒,同時比其他引發劑分解平穩,安全性高。改變AAPH 的濃度可以調節導致蛋白質氧化的過氧自由基的生成,以模擬蛋白質在長期儲存和運輸過程中的環境,并獲得類似于實際儲存和運輸的氧化蛋白質模型。因此,本研究以大豆蛋白為研究對象,采用AAPH 構建過氧自由基-大豆蛋白氧化體系,模擬工廠儲藏過程中實際產生的蛋白氧化聚集體,將蛋白氧化聚集體分別進行不同時間的微波改性處理,探究微波處理對蛋白結構特性和功能特性的影響,進而研究微波對大豆蛋白氧化聚集體結構的改變與功能特性變化的關系,以期從分子水平解析微波調控大豆蛋白氧化聚集體解聚作用機理,從而為提升大豆蛋白氧化聚集體的功能性質以及大豆蛋白產品的開發和儲藏提供理論基礎。
1 材料與方法
1.1 材料與試劑
大豆分離蛋白(SPIs 純度94.2%),山東禹王生態食業有限公司;偶氮二異丁脒鹽酸鹽 (AAPH) 美國Sigma 公司;十二烷基硫酸鈉(sodium dodecyl sulfate,SDS),上海阿拉丁生化科技股份有限公司;非轉基因一級大豆油,長春市遼都糧油有限公司;其他試劑均為國產分析純。
1.2 儀器與設備
M1-L213B 型微波爐,美的集團有限公司;pilot10-15EP 型真空干燥機,博醫康公司;Zetasizer Nano ZSP型納米粒度電位儀,馬爾文帕納科技有限公司;Nicolet is50 型傅里葉變換紅外光譜儀,賽默飛世爾科技有限公司;F-4 500 型熒光分光光度計,日本HITACHI 公司。
1.3 樣品制備
參考 CHENG 等[13]的方法,以10 mg/mL 的質量濃度把大豆分離蛋白溶解于磷酸鹽緩沖溶液(0.01 mol/L,pH 值7.2)中。避光條件下加入0.5 mmol/L 的AAPH 后恒溫37 ℃水浴處理6 h,然后使用14 000 kDa 透析袋4 ℃透析處理72 h,期間每隔4 h 更換一次去離子水,得到氧化大豆蛋白溶液,經過冷凍干燥后制得大豆蛋白氧化聚集體樣品,命名為OSPI。將制得的大豆蛋白氧化聚集體分別在功率為350 W 的條件下進行微波處理0、10、20、30、40、50、60、70 s 微波處理后制得7 種不同的微波處理大豆蛋白氧化聚集體樣品,按照微波處理時間的不同分別命名為WOSPI-10、WOSPI-20、WOSPI-30、WOSPI-40、WOSPI-50、WOSPI-60、WOSPI-70。
1.4 粒徑分布測定
將樣品用去離子水稀釋為質量濃度0.05 g/mL 的蛋白溶液,加入測量池中,采用納米粒度及Zeta 電位分析儀,設定參數為:蛋白質折射率等于1.460、分散劑折射率等于1.330,在(25±2) ℃下測定其粒徑分布特征和蛋白質分散指數。
1.5 濁度測定
參考文獻[14]的方法,并略加改動。將各組樣品采用磷酸鹽緩沖溶液配制成所需的蛋白濃度后,磁力攪拌60 min,采用熒光光度計測定600 nm 波長下的吸光度值A,濁度T由式T=1.032×計算獲得,式中T為濁度,A為吸光度值,V為稀釋倍數,I為光程(0.001 m)。
1.6 硫磺素T(Th T)熒光檢驗分析
將樣品用去離子水配制為10 mg/mL 質量濃度樣品溶液。將4 mg 的Th T 溶于250 mL 的pH 值7.2,0.01 mol/L 磷酸鹽緩沖溶液(含3 mmol/L 的NaCl),充分溶解后過0.22 μm 濾膜除去不溶物質,制得Th T 溶液。將50 μL的樣品溶液和5 mL 的Th T 溶液充分混勻后以Th T 溶液為對照,于440 nm 激發波長,482 nm 發射波長測定其熒光強度,狹縫寬度均為5 nm[15-16]。
1.7 傅里葉紅外掃描光譜分析
稱取2 mg 樣品與200 mg 溴化鉀研磨混勻壓片測定傅里葉紅外掃描光譜,測定溫度為25 ℃,設定參數如下:掃描波數為4 000~400 cm-1,波數精度為0.5 cm-1,分辨率為4 cm-1,掃描32 次。所得數據經Peak Fit 軟件對酰胺Ⅰ帶1 750~1 550 cm-1波段進行分析[17]。
1.8 內源性熒光光譜分析
使用RF-7 000 PC 熒光分光光度計測定樣品的熒光光譜,將樣品以1 mg/mL 的濃度溶于去離子水中得到樣品溶液,在激發波長290 nm、發射波長在300~400 nm、夾縫寬均為5.0 nm 的條件下測定樣品的內源性熒光光譜[18]。
1.9 溶解度測定
將樣品以1 mg/mL 的濃度溶于去離子水中,再于4 ℃下2 000 r/min 離心15 min 取上清液。以BSA 為標準蛋白,使用Lowry 法測定上清液中蛋白質的含量[19]。溶解度的計算公式為:
式中Ps為樣品蛋白質的溶解度,%;C上為上清液中的蛋白質含量,mg/mL;C為樣品中的蛋白質含量,取1 mg/mL。
1.10 持水性測定
參考MCCONNELL 等[20]的方法并略作修改。取300 mg樣品溶于30 mL 去離子水中。再于4 ℃下1 500 r/min 離心15 min 取沉淀,測量其質量。持水性的計算公式為:
式中WHC為樣品的持水性,%;W沉淀為沉淀的質量;W為樣品的質量,取300 mg。
1.11 持油性測定
參考LIN 等[21]的方法并略作修改。取100 mg 樣品溶于20 mL 大豆油中,于25 ℃漩渦振蕩15 min 使其充分混勻,再于4 ℃下1 500 r/min 離心15 min 取沉淀,測量其質量。持油性的計算公式為:
式中OHC為樣品的持油性,%;W沉淀為沉淀的質量;W為樣品的質量,取100 mg。
1.12 起泡性及泡沫穩定性
參考ZHANG 等[22]的方法并略作修改。將樣品以1 mg/mL的濃度溶于去離子水中,置于4 ℃冰箱中水化12 h 得到樣品溶液。取20 mL 樣品溶液置于100 mL 量筒內,使用IKA T18 ULTRA-TURRAX 型高速均質機以12 000 r/min的速度在室溫下均質2 min,記錄均質結束后0 min 及10 min時的泡沫高度(以量筒刻度為準)。起泡性及泡沫穩定性的計算公式為:
式中FC為樣品的起泡性,%;V0為樣品溶液均質結束后0 min 時的泡沫高度,mL;V為樣品溶液的體積,mL;FS為樣品的泡沫穩定性,%;V30為樣品溶液均質結束后30 min 時的泡沫高度,mL。
1.13 乳化活性和乳化穩定性測定
參考KINSELLA 等[23]的方法并略作修改。將樣品以1 mg/mL 的濃度溶于去離子水中,置于4 ℃冰箱中水化12 h 得到樣品溶液。取21 mL 的蛋白溶液與7 mL 的大豆油混合,使用IKA T18 ULTRA-TURRAX 型高速均質機以12 000 r/min 的速度在室溫下均質2 min 制得乳液。在乳液制備完成后的0 min 和10 min 時分別測定其吸光值。乳化活性和乳化穩定性的計算公式為:
式中EAI為樣品的乳化活性,m2/g;A0為乳液在0 min 時的吸光度;C蛋白為樣品溶液中的蛋白質含量,取0.001 g/mL;ESI為樣品的乳化穩定性,min:A10為乳液在10 min 時的吸光度;ΔT為兩次取樣的時間間隔,取10 min。
1.14 激光共聚焦顯微鏡分析
參考殷靜霖等[24]的方法并略作修改。先用異丙醇為溶劑制備1 mg/mL 的耐爾紅染料以及10 mg/mL 的耐爾藍染料,分別過0.22 μm 濾膜以去除不溶性物質。再取1.13 中制得的乳液100 μL 與700 μL 的去離子水混勻后,加入35 μL 的耐爾紅染料、40 μL 的耐爾藍染料。混勻后于避光環境靜置30 min 使其充分染色。使用染色的乳液制備激光共聚焦顯微鏡樣片,在550 nm 下激發耐爾紅,490 nm 下激發耐爾藍,于20×的物鏡觀察并拍攝。
1.15 數據處理與分析
所有的試驗重復3 次,結果用平均值±標準差表示,采用SPSS 19.0 軟件對試驗數據進行ANOVA 顯著性分析,P<0.05 為顯著性差異,采用Origin 9 軟件作圖。
2 結果與分析
2.1 微波對大豆蛋白氧化聚集體粒徑分布的影響
由蛋白質的粒徑分布能直觀地從宏觀角度反映物理場作用下蛋白質分子中發生的聚集與解聚現象[25]。微波處理對氧化大豆分離蛋白平均粒徑的影響見圖1。由圖1可知,與SPI 的粒徑分布相比,經過AAPH 氧化處理后的OSPI 粒徑分布變為三峰分布,原有粒徑峰均右移并在更大粒徑處形成一個新的峰。這是因為大豆分離蛋白的蛋白質骨架及側鏈基團受自由基攻擊而附加上側鏈基團,導致蛋白質發生了交聯聚集[26]。在微波處理后,WOSPI-10 的粒徑分布變為雙峰分布,當微波處理時間達到40 s 后WOSPI-40、WOSPI-50、WOSPI-60 的粒徑分布呈現出小粒徑峰降低,大粒徑峰右移的趨勢,在微波處理時間達到70 s 時,WOSPI-70 的雙峰均出現右移。這是因為短時間的微波處理能通過電場等非熱效應促進大豆分離蛋白分子運動,增加了分子間碰撞發生概率,使得大豆分離蛋白分子間的非共價鍵斷裂,原有的大粒徑的氧化聚集體分子解聚為小粒徑的氧化聚集體。而隨著微波處理時間的逐漸增加,大豆分離蛋白氧化聚集體碰撞加劇,系統內熱效應增強,誘導已經解聚的氧化聚集體分子出現變性,形成了新的化學鍵,增強了分子間相互作用力,繼而導致聚集體的出現,WOSPI 粒徑增加[27]。
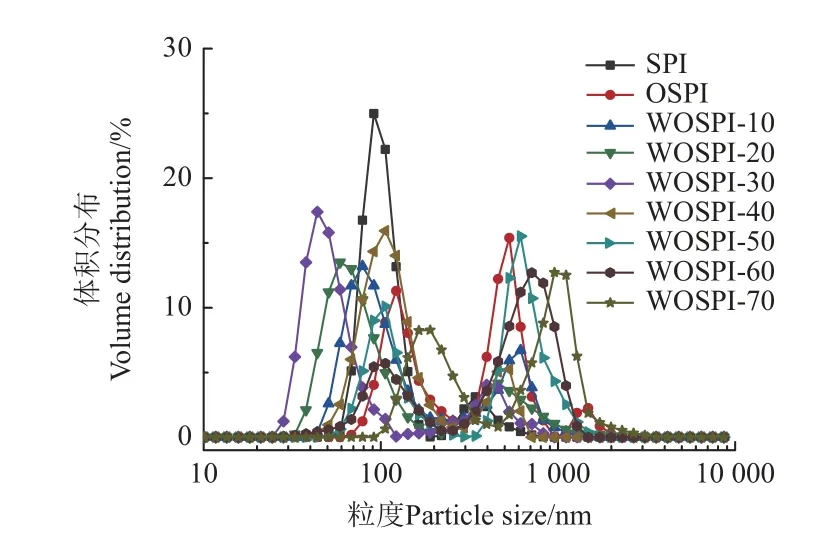
圖1 微波處理時間對大豆蛋白氧化聚集體粒徑分布的影響Fig.1 Effects of microwave treatment time on particle size distribution of OSPI (oxidized soybean protein aggregates)
2.2 微波對大豆蛋白氧化聚集體濁度的影響
蛋白質溶液的濁度取決于溶質即蛋白質自身的粒徑大小,兩者呈正相關,因此常用濁度來表征蛋白質的聚集和解離程度[28]。由圖2 可知,與SPI 的濁度相比,經過AAPH 氧化處理后的OSPI 的濁度顯著增加(P<0.05)。這表明氧化處理可能引起蛋白溶液中的大分子氧化聚集體含量升高和粒徑增加。隨著微波處理時間的增加,WOSPI 的濁度整體上先降低后增加,并在微波處理30 s時達到極小值。這是因為在短時的微波處理中,OSPI 主要受電磁場等非熱效應的影響,破壞了維持蛋白空間結構的非共價鍵,大豆分離蛋白分子間的聚合作用被削弱,導致WOSPI 的濁度在前期呈下降趨勢[29]。然而長時間的微波處理下,已經解離出來的小分子氧化聚集體與原有的大分子氧化聚集體的結構被熱效應破壞[30],暴露了蛋白質內部基團,增加了蛋白質分子間的聚合機會,形成了大粒徑的大豆蛋白聚集體,最終造成WOSPI 的濁度上升。

圖2 微波處理時間對大豆蛋白氧化聚集體濁度的影響Fig.2 Effects of microwave treatment time on turbidity of OSPI
2.3 微波對大豆蛋白氧化聚集體硫黃素T 熒光強度的影響
硫黃素T(Thioflavin T,Th T)能特異性地平行插入纖維狀蛋白聚集體內部的分子間反平行β-折疊并與其結合,其熒光強度可以用來表征蛋白質聚集程度的變化。由圖3 可知,與SPI 相比,OSPI 的Th T 熒光強度顯著升高,說明氧化處理顯著增加了大豆分離蛋白中分子間反平行β-折疊結構的含量。這可能是因為氧化處理會使得蛋白質分子通過二硫鍵等共價鍵的形成,誘導共價交聯,繼而通過分子間反平行β-折疊結構形成大分子的氧化聚集體。而經過微波處理后,WOSPI 的Th T 熒光強度隨微波處理的時間整體呈現先下降后增加的趨勢。這是因為微波處理引發的電場作用等非熱效應對大豆分離蛋白氧化聚集體內部的氫鍵產生了作用,部分氫鍵斷開,進而使得分子間反平行β-折疊結構含量減少[31]。當微波時間超過20 s 以后,大豆分離蛋白氧化聚集體發生變性,蛋白表面的疏水性基團等被修飾,在分子間形成了二硫鍵、疏水鍵等,誘導小分子聚集體交聯締合形成大分子的熱聚集體。而當微波處理時間達到70 s 時,Th T 熒光強度的下降可能是因為纖維狀聚集體的形成已經進入穩定期,生成速度減緩[32];同時熱效應過強,誘導進一步聚集團聚,包埋了部分分子間反平行β-折疊結構,使得Th T 無法與其結合,兩者共同導致了熒光強度的下降。
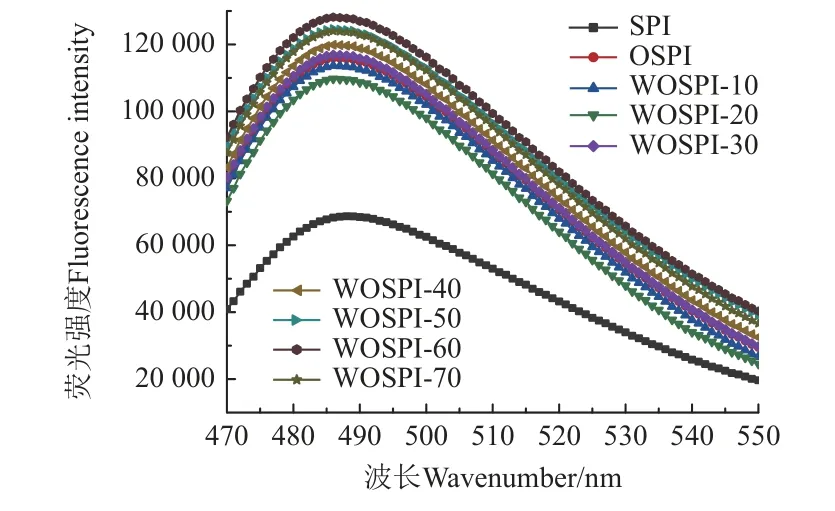
圖3 微波處理時間對氧化大豆分離蛋白硫黃素T 熒光強度的影響Fig.3 Effects of microwave treatment time on fluorescence intensity of Th T of OSPI
2.4 微波對大豆蛋白氧化聚集體二級結構的影響
由表1 可知,與SPI 相比,OSPI 的二級結構出現顯著變化(P<0.05),蛋白質有序結構的減少和無序結構的增加表明氧化處理能破壞蛋白原有結構,通過分子間反平行β-折疊形成氧化聚集體[33]。隨著微波處理時間的增加,WOSPI 的α-螺旋、β-轉角和β-折疊含量均呈現先增加后減少的趨勢,而無規則卷曲和分子間反平行β-折疊結構含量則相反。這是因為短時微波處理導致了大豆分離蛋白分子內部的偶極分子與極性側鏈間發生高頻振蕩,誘導部分氫鍵斷裂,增強了大豆分離蛋白的柔性,誘導了聚集體大分子的破碎。之后處理時間增加,存在的電場等非熱效應的存在使得分子間碰撞機率大,更容易發生相互作用連接在一起,加速了自組裝的過程,因而OSPI 內的無序結構再次增多。AKKERMANS 等[34]研究也表明,分子間的碰撞幾率增加會顯著促進蛋白聚集體的自組裝。但是當微波處理時間達到70 s 時,WOSPI-70 的分子間反平行β-折疊結構開始下降。這可能是因為分子間反平行β-折疊結構主要存在于聚集體核心中,而在微波處理帶來的熱效應作用下,聚集體形成進入穩定期,不再產生新的聚集體核心,反而轉為聚集體之間的進一步聚集[35]。
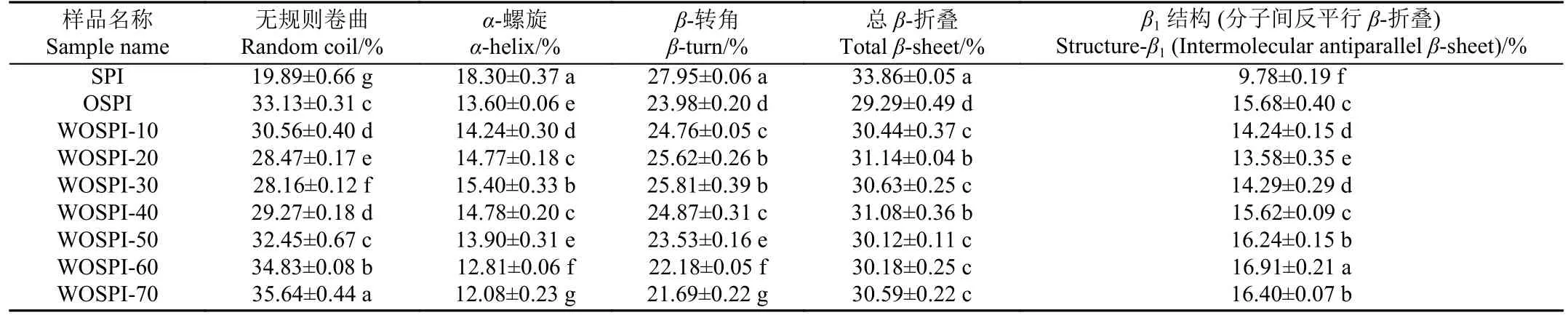
表1 微波處理時間對大豆蛋白氧化聚集體二級結構的影響Table 1 Effects of microwave treatment time on secondary structure of OSPI
2.5 微波對大豆蛋白氧化聚集體內源熒光強度的影響
由圖4 可知,與SPI 相比,OSPI 內源熒光強度顯著降低,這說明OSPI 中出現氧化聚集現象[36]。經過微波處理后,WOSPI 的內源熒光強度逐漸降低,最大吸收波長(λmax)先增加后減少,并在30 s 時達到極大值。這表明色氨酸殘基發生了熒光猝滅反應,且附近的微環境極性先增加后降低。這可能是因為微波處理導致的電場效應使得大豆分離蛋白分子間布朗運動加強,分子間碰撞增加,繼而處于激發態的大豆分離蛋白氧化聚集體分子間發生動態的熒光猝滅效應,導致熒光分子出現光度下降的現象;同時還會斷開大豆分離蛋白氧化聚集體內部的疏水鍵等非共價鍵,使得內部的色氨酸殘基暴露在極性環境中,進而導致λmax出現增加。處理時間過長時,大豆分離蛋白氧化聚集體重新交聯形成大分子的熱聚集體,色氨酸殘基被埋入熱聚集體的內部,繼而導致λmax降低。
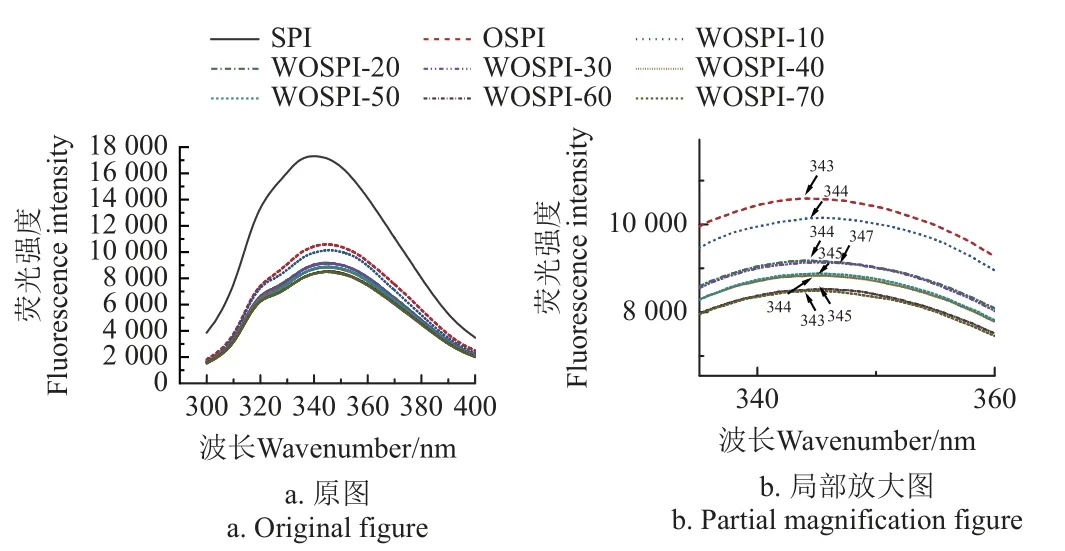
圖4 微波處理時間對大豆蛋白氧化聚集體內源熒光強度的影響Fig.4 Effects of microwave treatment time on endogenous fluorescence intensity of OSPI
2.6 微波對大豆蛋白氧化聚集體溶解度的影響
溶解度由蛋白質表面的疏水基團和親水基團的性質決定,與其起泡性、乳化性等功能性質密切相關[37]。由圖5 可知,與SPI 相比,OSPI 溶解度顯著降低(P<0.05)。這是因為在自由基在氧化過程中攻擊了大豆分離蛋白的主鏈和側鏈基團,形成了不溶性的氧化聚集體。隨著微波處理時間的增加,WOSPI 的溶解度呈現先增加后減少的趨勢。這是由于適當的微波處理可以通過其非熱效應使得蛋白分子極化,在水中高頻振蕩,非共價作用等被削弱導致結構更松散,從而使水分子容易進入蛋白內部與其發生水合,繼而導致溶解度增加。而微波處理時間進一步延長導致熱效應逐漸增強,大量暴露在大豆分離蛋白表面的疏水基團相互作用引發蛋白質發生締合,導致溶解度下降[38]。WANG 等[39]研究也表明,微波處理可以通過電場作用有效提升蛋白質的溶解度,但熱效應增強則會導致蛋白質聚集形成不溶性組分,進而導致溶解度下降。
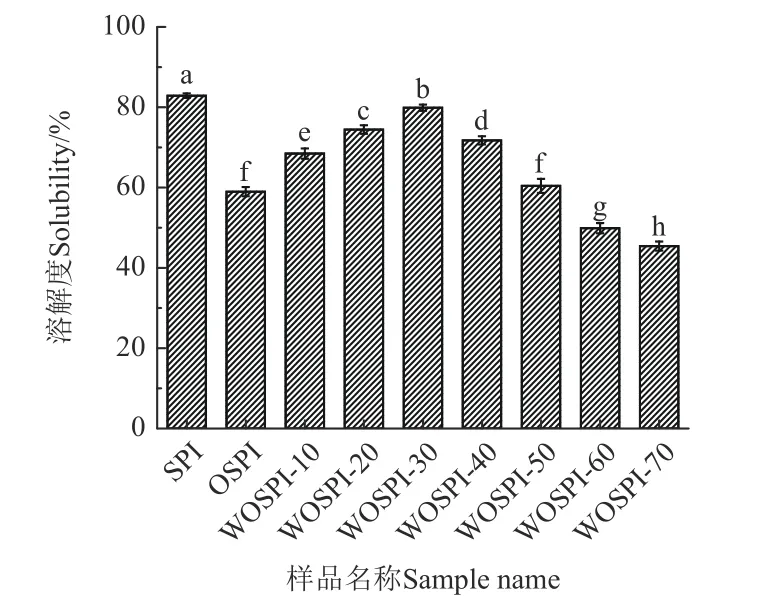
圖5 微波處理時間對大豆蛋白氧化聚集體溶解度的影響Fig.5 Effects of microwave treatment time on solubility of OSPI
2.7 微波對大豆蛋白氧化聚集體持水性的影響
持水性主要受蛋白質的構象等物理結構變化的影響[40]。由圖6 a 可知,與SPI 相比,經AAPH 氧化處理后的OSPI 持水性顯著下降(P<0.05)。這是因為氧化會導致蛋白質聚集,形成不溶性的氧化聚集體,使得大豆分離蛋白分子剛性增強,結構穩定性下降,導致持水性下降。微波處理導致的振蕩效應使得大豆分離蛋白分子內氫鍵、疏水鍵等非共價鍵斷裂,打開了團聚的聚集體結構,使得分子舒展,柔性結構舒張,可以有效地吸附水分子。同時大豆分離蛋白中的β-折疊結構有所增加,水分子更容易嵌入蛋白質空隙中,增強了大豆分離蛋白的持水性。之后微波處理時間延長,熱效應誘導小分子聚集體產生熱聚集,繼而形成大分子的不溶性熱聚集體,阻礙了與水分子的接觸,導致了持水性出現下降。鄧芝串等[41]研究表明,微波處理產生的熱效應逐漸增加,大豆蛋白隨之出現先展開后聚集的構象變化,持水性也出現了類似變化。
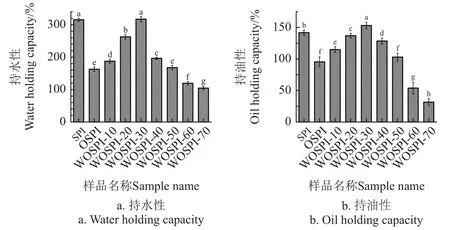
圖6 微波處理時間對大豆蛋白氧化聚集體持水性及持油性的影響Fig.6 Effects of microwave treatment time on water and oil holding capacity of OSPI
2.8 微波對大豆蛋白氧化聚集體持油性的影響
持油性是指一定重量的干基蛋白樣品對油脂的保持能力,其作用基礎來自蛋白質自身的疏水基團[42]。由圖6b可知,與SPI 相比,經過AAPH 氧化處理后的OSPI 的持油性顯著下降(P<0.05)。這是因為大豆分離蛋白被氧化形成了氧化聚集體,疏水親油性基團被包埋,阻礙OSPI與油脂分子結合。LI 等[43]研究也表明氧化會導致蛋白質表面疏水性的降低而降低持油性。隨著微波處理時間的增加,WOSPI 的持油性呈現出先增加后減少的趨勢,并在30 s 處達到極大值。這是因為短時微波處理中占據主導地位是非熱效應,促進大豆分離蛋白氧化聚集體碰撞導致部分解聚,分子內氫鍵、疏水鍵等非共價鍵斷裂,蛋白結構舒張,使得原本包埋在內部的疏水親油性基團暴露到蛋白表面,進而與更多的油脂吸附結合。但是隨著微波處理時間的進一步增加,表面的疏水性基團通過疏水相互作用發生交聯,同時游離巰基也互相結合形成了二硫鍵,進而形成了大分子的聚集體,疏水基團重新包埋分子內部,導致不能與油脂充分結合,降低持油性。
2.9 微波對大豆蛋白氧化聚集體起泡性及泡沫穩定性的影響
由表2 可知,與SPI 相比,OSPI 起泡性顯著下降,穩定性顯著增加(P<0.05)。這是因為氧化過程中的自由基攻擊大豆分離蛋白分子,導致蛋白變性,降低了蛋白柔性,形成了大分子蛋白氧化聚集體,不易在氣-水界面展開導致OSPI 起泡性降低[44-45]。經過微波處理后,OSPI 的起泡性隨著微波處理時間的增加呈現出先增加后減少的趨勢,泡沫穩定性整體來看也有相同趨勢,兩者分別于微波處理30 s 和20 s 處達到極大值。這是因為在微波的作用下,大豆分離蛋白出現極化現象,內部的非共價鍵被破壞,導致蛋白舒張展開柔性增強,促進了氣-水界面的形成;舒張后的蛋白質在界面膜上形成的網狀結構也更加穩定,所以增強了蛋白質的起泡性和泡沫穩定性。當處理時間過長時,熱聚集體的形成導致氣-水界面膜難以形成并維持,進而導致蛋白質的起泡性和泡沫穩定性出現下降。楊文敏等[46]研究表明,在適當微波處理巴旦木蛋白后,蛋白結構伸展,更容易和水分子結合,提高了蛋白的溶解度,促進了蛋白向氣-水界面擴散,繼而增強了蛋白的起泡性,這與溶解度的變化相一致。
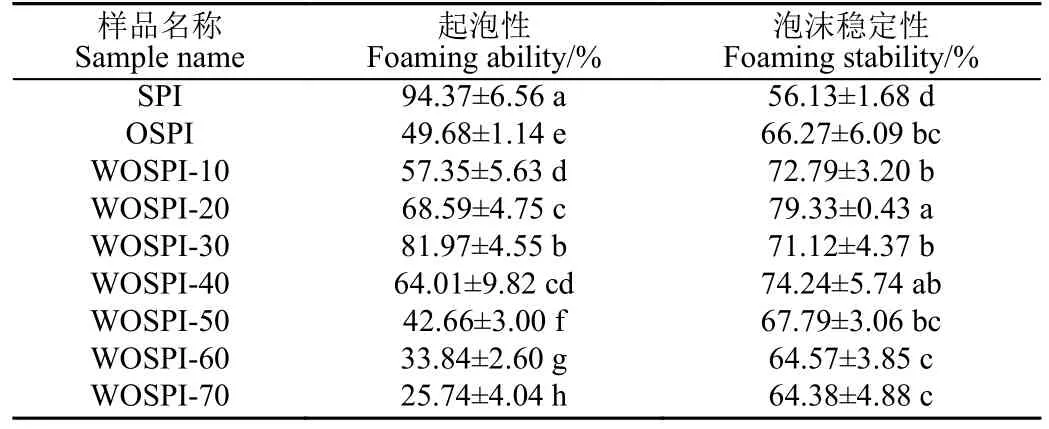
表2 微波處理時間對大豆蛋白氧化聚集體起泡性及泡沫穩定性的影響Table 2 Effects of microwave treatment time on foaming capacity and foaming stability of OSPI
2.10 微波對大豆蛋白氧化聚集體乳化活性和乳化穩定性的影響
由表3 可知,OSPI 的乳化活性和乳化穩定性比SPI顯著降低(P<0.05)。這可能是因為蛋白質氧化后降低了分子柔性,使得蛋白質與油-水界面難以結合,無法形成穩定的界面膜。隨著微波處理時間的增加,WOSPI 的乳化活性及穩定性均呈現先增加后降低的趨勢,分別在微波處理30、20 s 處達到極大值。研究表明,微波所產生剪切力和振蕩效應等導致氧化大豆蛋白聚集體的解聚和去折疊,蛋白質結構逐漸由有序變為無序,分子柔性增強,蛋白質的兩親性提高。因此,水-油界面的表面張力降低,從而增強了蛋白質的乳化特性。結合前面結果發現,較短時間內微波處理的非熱效應使得大豆分離蛋白出現極化現象,破壞了蛋白內部的非共價鍵,一些不穩定的氧化結構被打開,使得蛋白的剛性減弱,柔性增強;同時非共價鍵斷裂還導致了疏水性增高,促進油-水界面的形成,并誘導大豆分離蛋白更容易結合到油-水界面處吸附油脂并形成膜結構,提升其乳化能力。當微波處理時間繼續延長,微波處理產生的熱效應逐漸增強,暴露到蛋白質表面非極性基團增加,在非熱效應促進的分子碰撞中互相以疏水作用結合形成了更加致密的不溶性熱聚集體,阻礙了油-水界面的形成,降低了大豆分離蛋白在界面處的展開能力,進而導致乳化能力下降。
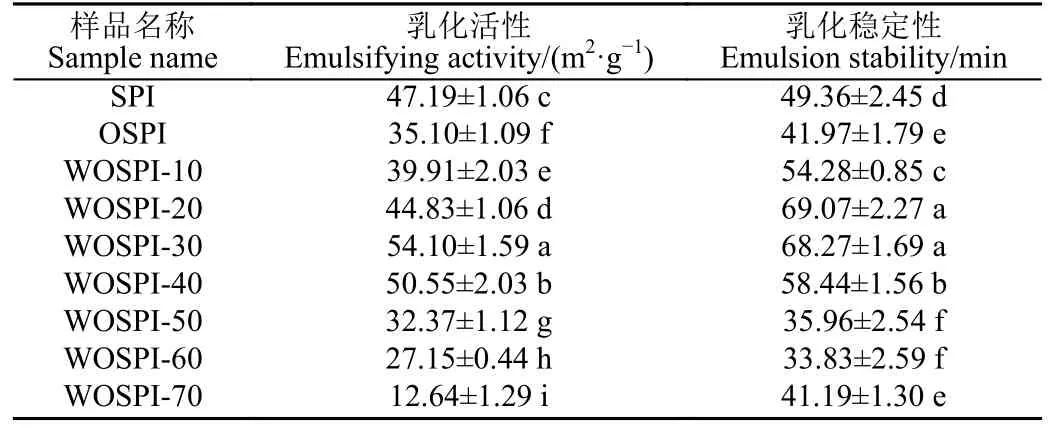
表3 微波處理時間對大豆蛋白氧化聚集體乳化活性及乳化穩定性的影響Table 3 Effects of microwave treatment on emulsifying activity and emulsion stability of OSPI
2.11 微波對大豆蛋白氧化聚集體乳液微觀形態的影響
使用尼羅藍和尼羅紅作為熒光染料分布對乳液中的蛋白質和油脂進行染色。由圖7 可知,未經微波處理的SPI制成的乳液液滴呈球形,整體分布較為均勻,而OSPI 制備的乳液粒徑顯著增加,出現了聚集現象。這是因為氧化導致大豆分離蛋白出現了聚集現象并形成了一定量的不溶性聚集體,影響了界面膜的形成和穩定性,降低了OSPI的乳化能力和乳液均一性。隨處理時間增加,WOSPI 制備的乳液液滴更小更均一,與粒徑結果相一致;當微波處理時間超過30 s 后,乳液液滴出現了粒徑增大的絮凝現象,乳液液滴逐漸失去圓形形態,變為不規則的大顆粒。這說明適當的微波處理可以通過振蕩等作用使得大分子的氧化聚集體破碎解聚成小分子聚集體,提升其乳化能力,幫助小分子蛋白聚集體吸附到油-水界面上并形成相對穩定的界面膜,進而提升了乳液的均一性。但是當微波處理時間超過30 s 后,WOSPI 乳液出現粒徑增大且逐漸聚集的趨勢。這說明乳化性正在降低,不能覆蓋原有的油-水界面,可能因為長時間微波處理帶來的熱效應導致了大豆分離蛋白間以二硫鍵、疏水鍵等形成聚集,繼而消耗并包埋了疏水基團,導致乳化能力降低,影響乳液穩定性和形成不規則的大分子乳液液滴。TENG 等[47]的研究表明,長時間微波處理帶來的強熱效應使得大豆蛋白形成了大分子聚集體,進而導致制備的乳液間也發生了一定的絮凝,形成了大小不一的乳液液滴。
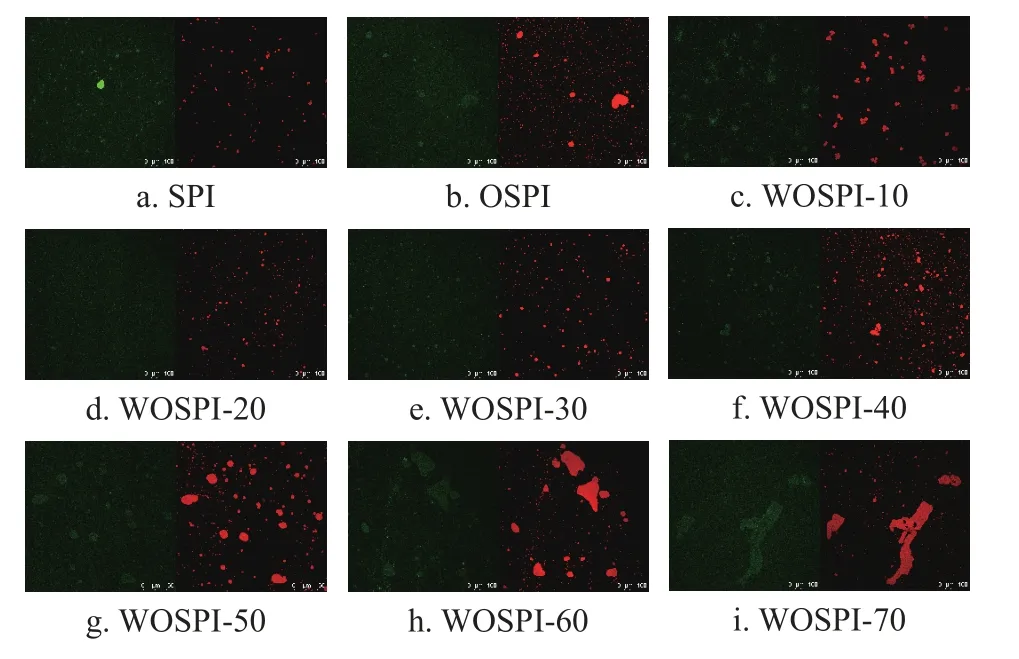
圖7 微波處理對大豆蛋白氧化聚集體乳液激光共聚焦顯微鏡圖的影響Fig.7 Effects of microwave treatment on the picture of OSPI emulsion by CLSM
3 結論
本研究以大豆蛋白為原料,采用偶氮二異丁脒鹽酸鹽(2,2′-azobis (2-amidinopropane) dihydrochloride,AAPH)制備大豆蛋白氧化聚集體,然后探討不同時間的微波處理技術對其結構和功能特性的影響。結果表明:氧化處理破壞大豆蛋白的結構,暴露其疏水性基團,并通過疏水相互作用形成二硫鍵,引起粒徑和濁度增大,形成大分子氧化聚集體,進而導致功能活性下降;短時間(<30 s)的微波處理可以促進大豆蛋白氧化聚集體發生解聚,引起粒徑和濁度減小,增強了分子柔性,無序結構減少,同時誘導蛋白結構舒張展開,包埋在蛋白內部的疏水基團和帶電基團暴露,結合位點增多,進而導致持水性,持油性提高,增強了蛋白的界面性質,導致起泡性和乳化活性的提高;而長時間(>30 s)的微波處理帶來的熱效應增加,會導致解聚的氧化聚集體通過共價和非共價相互作用形成二硫鍵,包埋了表面的疏水性基團和帶電基團并形成了致密的大分子熱聚集體,阻礙了水分子和油脂分子與大豆蛋白氧化聚集體的結合,導致其持水性、持油性下降,這也對大豆蛋白氧化聚集體的乳化性質、泡沫性質產生了一定的負面影響,可為今后的研究中繼續探討微波作用中熱效應和非熱效應分別對于大豆蛋白的影響提供一定的理論基礎。本研究結果闡明了不同微波處理時間對大豆蛋白氧化聚集體功能性質及結構的影響,為大豆蛋白氧化聚集體功能性質的改善及微波在氧化聚集體行為調控的應用方面提供參考。
猜你喜歡
農業科技通訊(2023年1期)2023-02-12 07:09:18
今日農業(2022年16期)2022-11-09 23:18:44
中國化肥信息(2022年7期)2022-08-31 01:29:28
中國化肥信息(2022年5期)2022-08-30 01:58:26
今日農業(2021年20期)2021-11-26 01:23:56
今日農業(2021年14期)2021-10-14 08:35:34
下一代英才(酷炫少年)(2018年6期)2018-07-09 03:17:44
農產品市場周刊(2017年4期)2017-03-03 19:40:05
兒童故事畫報·智力大王(2015年10期)2016-01-27 01:01:35
讀寫算(中)(2015年10期)2015-11-07 07:24:12
