SLC6A1基因變異導致神經發育障礙的家系分析
2023-08-10 05:08:48陳麗卿付俊一蘇堂楓
中風與神經疾病雜志 2023年4期
陳麗卿, 付俊一, 蘇堂楓, 劉 艷
SLC6A1基因(MIM 137165)定位于染色體3p25.3,在人類和其他動物的發育期與成熟大腦中均廣泛表達。SLC6A1基因編碼γ-氨基丁酸轉運蛋白1(gamma-aminobutyric transporter 1,GAT-1),該蛋白負責突觸γ-氨基丁酸(gamma-aminobutyric,GABA)的再攝取和細胞外間隙清除[1,2]。當SLC6A1基因發生致病性突變時,GAT-1功能喪失,影響突觸前膜從細胞外再攝取GABA,從而誘發癲癇。目前國內SLC6A1基因變異導致的癲癇報道少,主要報道的癲癇發作類型為肌陣攣-失張力發作及失神發作,且都為散發病例[3,4],我們報道1組SLC6A1 基因雜合變異引起癲癇的家系。
1 對象與方法
1.1 對象 先證者,女,于2歲6月齡就診于我科門診,系第1胎第1產,足月順產,出生無窒息史。4月齡抬頭,8月齡獨坐,1歲8月齡獨行。獨立行走后出現腿抖,伴有頻繁眨眼,獨站不穩及摔跤,無腿抖時可以獨行。就診時會說2個字。于2歲3月齡時曾行康復訓練,仍有間斷腿抖,摔跤,不敢行走。語言無進步。查體:體重12 kg,頭圍49 cm。面容無特殊,四肢肌力、肌張力正常,病理反射未引出。指鼻試驗不配合。可扶行,叫名無回應。血肝腎功能、電解質、乳酸、丙酮酸、血氨、空腹血糖、血尿代謝篩查正常。視覺及聽力誘發電位正常。視頻腦電圖(video electroencephalogram,VEEG):背景節律慢化(δ、θ混合慢波);睡/醒期見雙側前頭部、后頭部或廣泛性高波幅慢波、棘慢波或節律發放(見圖1)。頭部磁共振(magnetic resonance imaging,MRI):雙側額葉斑點狀高T2Flair信號灶,少許脫髓鞘病變可能(見圖2);左側顳角較右側大。Gesell嬰幼兒發育量表:一般發育水平約21個月,各項發育落后,發育商64。兒童孤獨癥評定量表提示無明顯孤獨癥表現。

圖1 VEEG:背景節律慢化(δ、θ混合慢波);睡/醒期見雙側前頭部、后頭部或廣泛性高波幅慢波、棘慢波或節律發放

圖2 頭部MRI:雙側額葉斑點狀高T2 Flair信號灶,少許脫髓鞘病變可能
家族史:基因結果出來后追問病史獲知患兒母親年幼時有癲癇發作,曾口服藥物治療1年(具體不詳)。目前無發作,但反應稍遲鈍。患兒外婆幼時有抽搐病史,用藥不詳,目前無發作。
1.2 方 法
1.2.1 家系全外顯子組測序 經監護人簽署知情同意書,并經醫院倫理委員會批準后,抽取患兒及父母,外婆外周血3 ml,提取DNA。應用 xGen Exome Research Panel v1.0 進行目標區域捕獲,使用illumina Hiseq X-ten 二代測序儀對富集的文庫進行 PE150 測序,平均測序深度為 100X,95%以上的區域達到 20X 以上的覆蓋度。
1.2.2 多重連接探針擴增技術(multiplex ligation-dependent probe amplification,MLPA)檢測 使用甲基化特異性的MLPA試劑盒(荷蘭MRC-Holland公司,ME028),檢測15q11.2-q13區域印記區缺陷、甲基化及單親二倍體。
1.2.3 Sanger測序 應用Sanger測序對篩查到的變異進行驗證,同時對其父母及外婆的相同基因位點進行驗證。
1.2.4 文獻復習 以“SLC6A1”、“癲癇”及“SLC6A1”、“epilepsy”為關鍵詞,對萬方數據庫、中國知網和Pubmed建庫至2023年1月收錄的論文進行檢索,并總結攜帶SLC6A1基因突變的國內患兒的臨床表型和基因突變特點。
2 結 果
2.1 治療及隨訪 患兒于2歲6月齡加用丙戊酸治療(11 mg/kg·次,bid),隨訪至2022年10月,有20個月無發作,可獨立行走、奔跑,語言障礙較前明顯好轉。
2.2 全外顯子測序結果 發現先證者存在SLC6A1基因c. 889G>A(p. Gly297Arg)的雜合變異,屬于常染色體顯性遺傳,此變異來自母親及外婆(見圖3)。已有文獻報道過該變異位點,但Carvill等[5]報道的攜帶該變異位點的患兒有重度智力障礙,自閉癥特征、攻擊行為、脊柱側彎、多種癲癇發作形式及對多種抗癲癇發作藥物療效差。Aguilera等[6]報道的患兒與本例相似,臨床表型相對要輕,表現為Angelman樣綜合征,且丙戊酸單藥控制良好。Piniella等[7]通過在過表達SLC6A1基因Gly297Arg突變體的人胚胎腎細胞293(human embryonic kidney 293,HEK293)中檢測GABA攝取活性,發現與野生型(Wide-type,WT)相比,其攝取活性顯著下降(低于WT的5%),證明該變異為致病性變異。

圖3 家系全外顯子組測序:先證者存在SLC6A1基因c. 889G>A(p. Gly297Arg)的雜合變異。此變異來自母親及外婆
2.3 MLPA檢測 本例患兒發作間期2次腦電圖均表現為背景節律慢化(δ、θ混合慢波),醒睡期均可見雙側前頭部、后頭部或廣泛性高波幅慢波、棘慢波,與Angelman綜合征患兒腦電圖表現相似,且患兒有全面性發育遲緩、言語障礙及刻板動作,但患兒并無Angelman綜合征快樂面容。所以本例基因檢測方法包括MLPA檢測,排除Angelman綜合征可能。結果染色體15q11-q13區域MLPA檢測顯示,在試劑盒檢測范圍內拷貝數和甲基化位點未見異常。
2.4 Sanger測序驗證 先證者SLC6A1基因存在c. 889G>A(p. Gly297Arg)的雜合變異。先證者母親及外婆均存在同樣的雜合變異(見圖3)。
2.5 文獻復習 以“SLC6A1”“癲癇”以及“SLC6A1”“epilepsy”為關鍵詞,對萬方數據庫、中國知網、Pubmed建庫至2023年1月收錄的論文進行檢索,共檢索到4篇[3,4,8,9]關于國內患兒攜帶SLC6A1基因變異的報告。結合本例患兒,目前國內共報道8例(男5例,女3例)SLC6A1突變的患兒(見表1)。8例患兒均有癲癇發作,起病年齡1~3歲。發作類型包括失神發作(5例,其中1例為不典型失神),肌陣攣發作(6例),肌陣攣-失張力發作(5例),全面強直陣攣發作(1例)。腦電圖均可見廣泛性棘慢波、多棘慢波發放,2例患者背景節律正常。8例患兒均存在發育遲緩,語言發育落后是突出特征。2例患兒報道存在自閉癥樣表現,1例患兒報道存在刻板樣動作,1例患兒報道存在注意缺陷多動障礙。頭部核磁檢查無特異性異常表現。所有患兒均加用丙戊酸抗癲癇治療,7例患兒對抗癲癇藥反應良好,6例隨訪后期無發作,1例發作次數明顯減少。1例患兒表現為難治性癲癇。除本例患兒SLC6A1基因存在家系遺傳,其余患兒均為新發突變。1例為剪切變異,2例為無義變異,1例為缺失變異,其余4例均為錯義變異。1例存在SLC6A1基因缺失變異的患兒還存在NOTCH1和PRIMPOL基因變異,所以該患者還具有主動脈瓣狹窄和高度近視的表現。
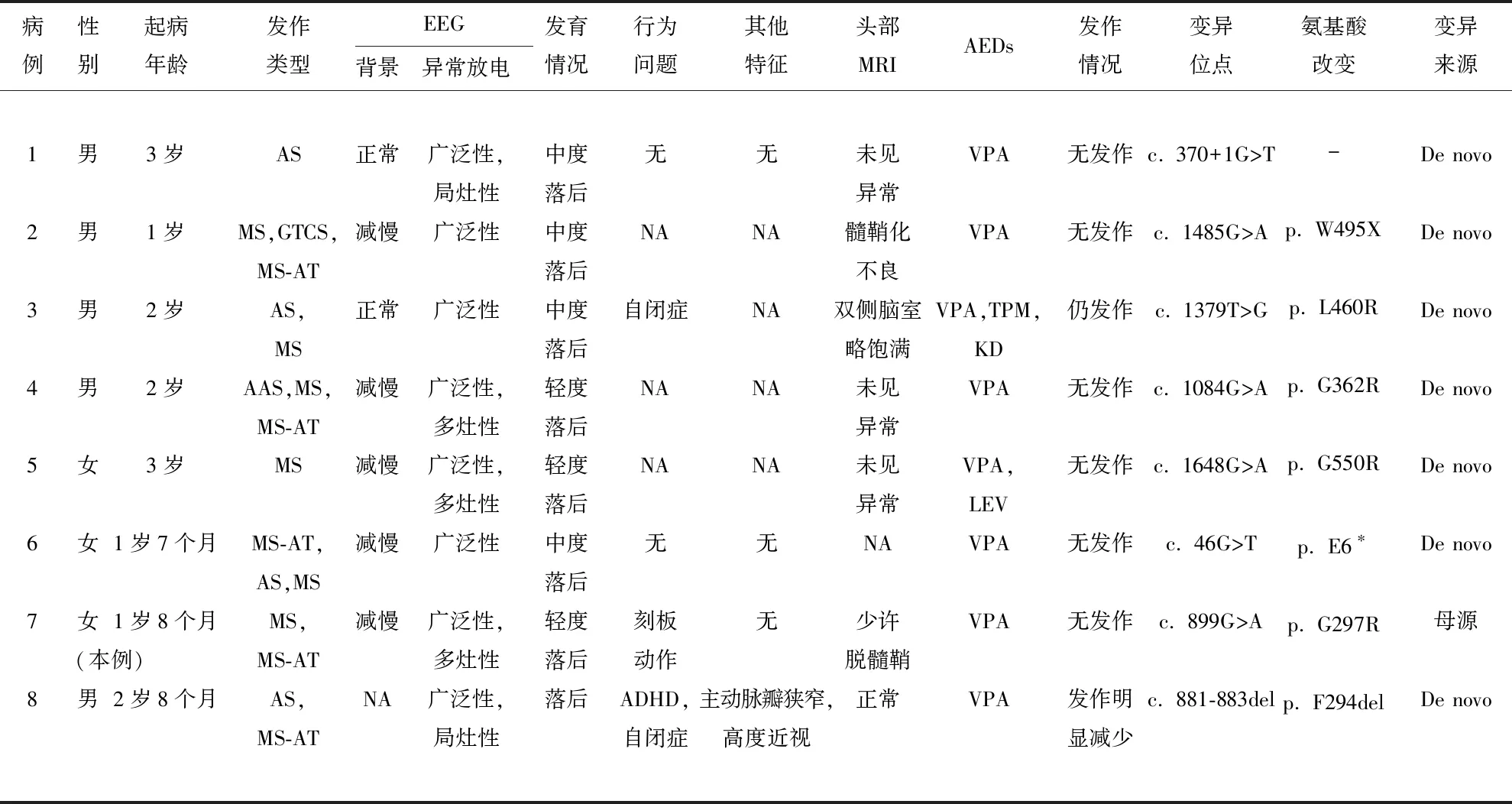
表1 8例SLC6A1基因變異的國內患兒臨床特征及基因型
3 討 論
SLC6A1相關疾病在2015年被首次描述[5]。SLC6A1 基因包含16個外顯子,長度約46.5 kb,其編碼的GABA 轉運體 GAT-1 主要位于 GABA 能中間神經元的軸突和神經終端,GAT-1可從突觸間隙重攝取GABA。GABA是哺乳動物中樞神經系統重要的抑制性神經遞質,調節抑制性突觸傳遞作用,從而防止腦內神經元過度興奮[10]。目前研究表明,SLC6A1基因變異,可引起多種神經發育障礙。Goodspeed等總結了116例SLC6A1基因變異患者的臨床資料,發現癲癇(91.1%)、發育遲緩及認知障礙(82.1%)、自閉癥(22.8%)是最常見的臨床特征[11]。
在一項隊列研究中,采用高通量測序(next generation sequencing,NGS)聯合拷貝數變異,對9 769例兒童癲癇患者進行篩查,發現SLC6A1被列為致病變異數較多的前10~20個基因之一[12]。
Johannesen等[13]報道16例SLC6A1基因突變患者,13例為點突變、3例為缺失突變,臨床表現為癲癇2 例(發作形式包括肌陣攣發作、失神發作、非驚厥性癲癇持續狀態、失張力發作、全面性強直陣攣發作、肌陣攣失張力發作),智力障礙16例(輕度智力障礙患者 11 例、中度智力障礙患者 4 例、1 例為嚴重的智力障礙),語言發育遲滯2 例,孤獨癥7例,注意力不集中2例等。但國內報道的8例患兒均有癲癇發作和發育遲緩,且語言發育遲滯是突出特點。
目前認為SLC6A1基因突變是發育性和癲癇性腦病的原因之一。據報道[11],癲癇首次發作的平均年齡是2.5歲。最常見的癲癇綜合征是早發性癲癇伴肌陣攣失張力發作(20/82,24.3%),其次是遺傳性全面性癲癇(19/82,23.1%)和非獲得性局灶性癲癇(8/82,9.75%)。在56例獲得詳細癲癇發作形式的患者中,最常見的是不典型失神發作(71.7%)、失張力發作(44.4%)和肌陣攣發作(27.7%)。廣泛性癲癇樣放電(36/52,69.2%)是最常見的腦電圖異常,可伴有背景節律變慢。這些特征與國內病例相似。但是對SLC6A1相關疾病而無癲癇發作的個體的腦電圖特征目前知之甚少。本例患兒的腦電圖特征是背景節律慢化,醒睡期可見雙側前頭部、后頭部或廣泛性高波幅慢波、棘慢波,與Angelman綜合征患兒腦電圖表現相似,雖然患兒并無Angelman綜合征的快樂面容,但考慮到患兒有全面性發育遲緩、言語障礙及刻板動作,所以本例基因檢測方法包括MLPA檢測,排除Angelman綜合征可能。根據Aguilera等的報道[6],通過全外顯子測序技術對14例Angelman樣綜合征患兒進行檢測,發現了10種可引起Angelman樣綜合征的新的基因,其中之一就包括SLC6A1基因,且突變位點和本例患兒相同。Angelman綜合征是母源UBE3A基因功能異常導致的神經發育遺傳性疾病。Aguilera等認為,SLC6A1基因變異出現Angelman樣綜合征,可能由于SLC6A1基因受到UBE3A基因的調控。但文獻中更多的關注基因型與表型之間的關系,對于腦電圖特征并未進行描述。
在一項較大的關于自閉癥測序的研究中(n=11 986),與23598個健康對照者相比,SLC6A1是自閉癥患者變異較豐富的前10個基因之一[14]。有學者總結了27 例SLC6A1基因變異患兒的基因型與臨床表型,18 例起病前有發育遲緩,11 例起病后出現發育倒退,9 例有自閉癥表現,100%病例均遺留不同程度的智力障礙(intellectual disability,ID),語言障礙是最常見的特征[3]。本例患兒有明顯語言障礙,雖有一些刻板動作,但自閉癥相關量表評估未達到診斷標準。且國內病例僅有2例報道有自閉癥樣表現。
Goodspeed等[11]總結的116例SLC6A1基因變異患兒中發現85 種變異,變異類型包括88個錯義變異,15個導致功能完全喪失的蛋白質截斷變異,7個剪接位點的變異,3個拷貝數大的缺失變異,2個小的插入和缺失和1個同義變異,其中錯義變異最常見(75.8%)。本例患兒為SLC6A1基因c.889G>A(p.Gly297Arg)變異為錯義變異,既往已有文獻報道[5~7],但雖為同一位點突變,既往報道的患兒臨床表型嚴重程度與本例有差異,Carvill等[5]報道的患兒臨床表現為重度智力障礙,自閉癥特征、攻擊行為、脊柱側彎、多種癲癇發作形式及對多種抗發作藥物療效差。最近的一些研究已經證明不同類型的變異對GAT-1活性的影響程度不同,比如截斷變異可能導致單倍劑量不足從而降低GAT-1表達,而一些錯義突變可能通過顯性-陰性效應導致GAT-1表達更低[15]。Piniella等[7]的研究結果顯示,本例患兒攜帶的Gly297Arg變異對細胞內運輸的影響是高度有害的,且該變異的GAT-1被保留在內質網中,完全無活性。這些研究表明,突變的GAT-1在內質網中的保留和降解會導致經過正確折疊、翻譯、修飾的功能性GAT-1表達降低[16]。但這些仍不能解釋即使是同一種變異,其臨床表型也有差異,基因型和表型的相關性,還需進一步的深入研究。
目前還沒有針對SLC6A1相關疾病的精準治療藥物,大多數患者需多學科聯合治療,包括神經病學、發育兒科、遺傳學以及言語和職業治療,以進行綜合管理[17,18]。根據之前研究,丙戊酸是最有效的抗癲癇藥物,拉莫三嗪和乙琥胺也顯示效果良好[19],此外,其他調節GABA濃度的抗癲癇藥,如氨己烯酸和噻加賓也可能發揮作用[20]。本例患兒采用丙戊酸單藥治療即有效,且患兒母親及外婆僅幼時有癲癇發作,抗癲癇藥治療效果良好。但仍有許多SLC6A1基因變異患者表現為難治性癲癇。Mermer等[15,16,21]研究發現,用4-苯基丁酸(phenylbutyrate,PBA)處理攜帶致病性SLC6A1變異的細胞,可增加GAT-1的活性。這可能是由于PBA作為伴侶蛋白,減少了突變的GAT-1在內質網中的保留,并增加了功能性GAT-1的表達。這項研究可能會對藥物治療SLC6A1相關疾病做出進一步貢獻。
綜上所述,本研究中SLC6A1基因變異患兒主要表現為幼兒期起病的肌陣攣失張力癲癇,同時伴有輕度的認知障礙及語言障礙,藥物治療后無發作。患兒母親及外婆臨床表型較患兒輕,僅有幼時癲癇發作,而無明顯認知障礙。通過文獻復習,SLC6A1基因變異可導致多種神經發育障礙臨床表型,可表現為癲癇(失神、失張力、肌陣攣及肌陣攣-失張力發作為主)、發育遲緩、認知障礙、孤獨癥或孤獨癥樣表現及注意缺陷多動障礙等。SLC6A1基因突變可以為錯義變異、無義變異、移碼變異、剪切變異、染色體微缺失等。基因型與表型之間的關系還需進一步研究。化學伴侶蛋白PBA是治療SLC6A1相關疾病的潛在有效藥物。
猜你喜歡
中國民間療法(2021年5期)2021-06-09 09:21:04
文萃報·周五版(2021年14期)2021-06-08 22:33:19
科教新報(2021年14期)2021-05-11 05:47:00
中國生殖健康(2020年7期)2021-01-18 03:02:02
海峽姐妹(2017年5期)2017-06-05 08:53:17
飲食科學(2017年5期)2017-05-20 17:11:53
絲路藝術(2017年6期)2017-04-18 13:59:02
丹青少年(2017年2期)2017-02-26 09:11:05
安徽醫科大學學報(2015年9期)2015-12-16 11:09:44
西南軍醫(2015年4期)2015-01-23 01:19:30
