僅腦脊液抗體陽性的髓鞘少突膠質細胞糖蛋白抗體相關疾病1例報告及文獻復習
2023-08-10 05:08:54裴曉晨秦毅丹陳加俊
中風與神經疾病雜志 2023年4期
裴曉晨, 李 佳, 秦毅丹, 蘇 杭, 陳加俊
髓鞘少突膠質細胞糖蛋白抗體相關疾病(MOG antibody disease,MOG-AD)是中樞神經系統的一種炎癥性脫髓鞘疾病,臨床特征為單相或復發相的神經功能障礙,此類疾病不符合多發性硬化及其他已知神經炎癥疾病的典型標準。多數MOG-AD患者血清中MOG抗體呈陽性、腦脊液中為陰性。目前僅有少數報道該疾病患者MOG抗體僅在腦脊液中呈陽性。然而,我們在臨床中發現1例少見的MOG-AD患者,該患者腦脊液中MOG抗體為弱陽性(1∶10),血清中抗體為陰性,結合影像及其他相關輔助檢查,考慮該患者免疫反應始發于顱內的膠質細胞等部位,由硬腦膜竇中的抗原提呈細胞(APC)捕獲腦脊液中的MOG抗原后產生一系列免疫反應,從而產生MOG抗體[1]。僅在腦脊液中MOG抗體陽性的患者與外周血中MOG陽性、腦脊液中MOG陽性/陰性的MOG-AD患者相比,可能血腦屏障未遭受破壞,提示免疫反應可能僅發生在顱內,具體機制目前尚未明確。該患者接受糖皮質激素沖擊治療后,逐漸遞減激素劑量直至停藥,患者腦脊液MOG抗體轉陰,病灶明顯減小,患者現恢復良好,未出現復發。
1 病例資料
患者,女,51歲,入院1年前在無明顯誘因的情況下開始出現飲水嗆咳及小便失禁,入院15 d前自覺右側肢體麻木,表現為“蟻走感”;12 d前出現右下肢無力,逐漸加重,無法獨立行走,而后相繼出現右上肢無力,不能持物;3 d前自覺氣短,說話時需深呼吸,患者上述癥狀逐漸加重,來吉林大學中日聯誼醫院診治。患者病程中伴有言語不清、飲水返嗆、左側頭痛、大便費力、有時小便失禁。該患者否認相關遺傳疾病的家族病史。
1.1 入院時神經系統查體 意識清醒,構音障礙,左側咽反射遲鈍,右側肢體肌力3級,左側肢體肌力5級,四肢腱反射活躍,四肢肌容積正常,雙側指鼻試驗不穩準,雙側快速輪替試驗笨拙、雙側跟膝脛試驗不穩準,右側肢體痛溫覺、位置覺減退,雙側Babinski、Chaddock征陽性,余神經系統查體未見異常。
1.2 影像學檢查結果 患者磁共振成像(MRI)檢查結果顯示,該患者病灶表現為延髓內邊界模糊的斑片狀異常信號影,中腦也存在可疑改變,病變性質考慮為脫髓鞘病變可能性大,待除外占位。(A)~(G)為入院時MR結果,病灶面積較大、信號較高;(H)~(I)為治療12 d后復查MR結果,病灶面積縮小、信號減低(見圖1)。
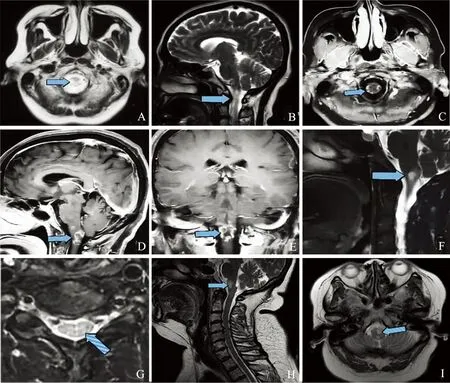
圖A、B 頭部MR平掃+彌散提示:延髓見斑片狀異常信號影,T1WI呈等、低信號,T2WI及FLAIRA呈高信號,DWI未見受限,T2WI矢狀位似見中腦少許稍高信號影;圖C~E 磁共振頭部普通增強提示:延髓見結節狀、斑片狀異常信號影,呈不規則環形強化,大小約(0.5~1.8)×1.0 cm;圖F、G 頸椎MR平掃提示:延髓內見斑片狀稍長T1,稍長T2異常信號,壓脂像呈稍高信號。頸4-5間盤水平硬膜囊后緣見低信號壓跡;圖H 頸椎MR平掃提示:延髓內見斑片狀稍長T1,稍長T2異常信號,壓脂像呈稍高信號;圖I 頭部MR平掃提示:延髓內可見不規則混雜長T1混雜長T2信號影,FLAIR呈高信號。

1.3 入院后病情變化 患者入院后第2天呼吸困難加重,主要表現為氣短、呼吸無力。患者在低流量吸氧狀態下血氧飽和度維持在98%,但咳嗽力量尚可,血氣分析:SO298%。結合患者病史、癥狀特點及目前輔助檢查,考慮患者延髓及高頸段病變為脫髓鞘可能性大,但不能除外為延髓腫瘤性病變(見圖1A~G)。神經外科會診后考慮患者病變位置特殊,無法進行活檢及手術治療。因此與患者及家屬溝通后,考慮暫時按照脫髓鞘病變進行治療,給予患者甲潑尼龍靜脈滴注治療(1000 mg·d-1,持續3 d,每3 d減半)。與此同時,進行視神經誘發電位檢查:VEP:兩眼分別刺激,雙側各波波形分化可,雙側N75、P100、N145潛伏期正常,波幅正常。提示:雙側視覺徑路傳導未見異常,患者視力一直未受影響。并完善腰椎穿刺術,腦脊液為無色清亮液體,測得腦脊液壓力90 mmH2O,腦脊液常規、生化提示腦脊液白細胞計數 14×106/L。血常規、凝血四項、肝功腎功、貧血兩項、同型半胱氨酸未見明顯異常。隨后化驗血清及腦脊液多發性硬化、視神經脊髓炎等相關抗體指標。結果提示,腦脊液中MOG抗體為弱陽性(1∶10),血清中抗體均為陰性(見圖2、圖3)。患者使用甲潑尼龍治療后病情逐漸緩解,2 d后呼吸困難、右側肢體麻木、無力逐漸有所好轉。

腦脊液的起始稀釋梯度為1∶1,血清的起始稀釋梯度為1∶10,加號數量代表該稀釋度下的熒光強度;通過基于細胞轉染的間接免疫熒光法(cell based assay,CBA)進行檢測。
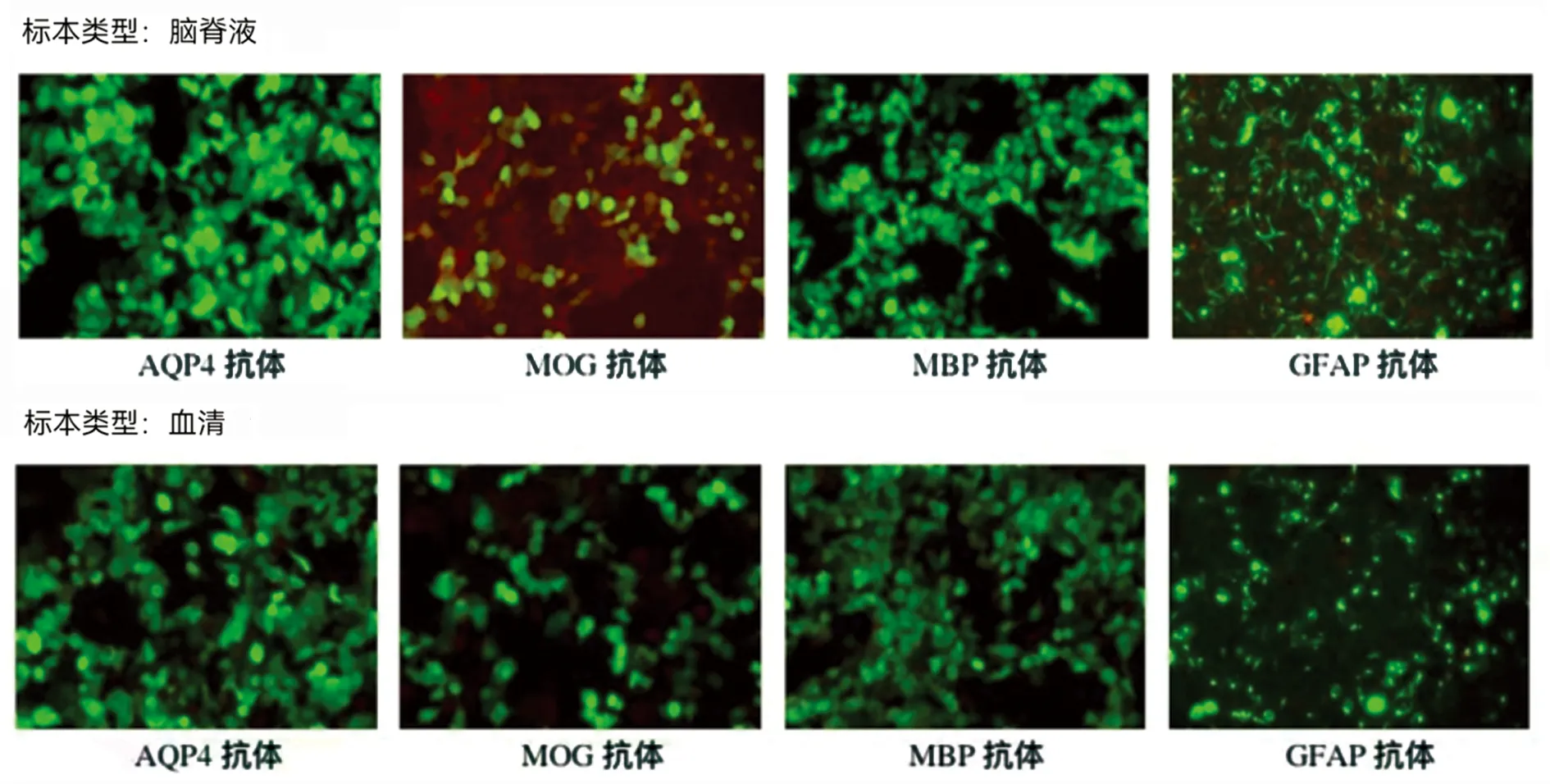
圖3 神經免疫檢測熒光圖片
使用糖皮質激素治療后12 d,復查腦脊液、血清相關抗體均為陰性,腦脊液常規生化提示腦脊液糖 4.1 mmol/L。復查頭部MR提示病灶范圍略有縮小(見圖1H、圖1I)。且患者右側上下肢肌力有明顯改善,出院時肌力4級+。出院后繼續規律口服潑尼松龍,無言語不清、肢體麻木及無力。出院4個月,患者復查頭部MR提示病灶范圍較前縮小,患者恢復良好,暫未出現復發。
2 討 論
髓鞘少突膠質細胞糖蛋白(MOG)是少突膠質細胞產生的位于髓鞘表面上的蛋白質之一,少突膠質細胞是中樞神經系統中的形成髓鞘的細胞,MOG是少突膠質細胞膜表面的重要組成部分;在髓鞘的形成,維持和分解中具有基本作用,且MOG只在中樞神經系統被發現,可以起到細胞黏附分子的作用,參與調節少突膠質細胞微管穩定性并介導補體級聯反應,通過MOG的黏附功能和髓鞘與免疫系統之間相互作用的維持髓鞘的結構完整性[2,3]。
近年來,已經證實,一些血清水通道蛋白4(AQP4-IgG)抗體陰性的視神經脊髓炎譜系疾病(NMOSD)患者具有抗髓鞘少突膠質糖蛋白(MOG)的IgG抗體,被診斷為MOG-AD[MOG-IgG的相關性腦脊髓炎(MOG-EM)或MOG-IgG的自身免疫病],是一種中樞神經系統(CNS)炎性脫髓鞘疾病,不同于多發性硬化癥(MS)和血清AQP4-IgG陽性神經脊髓炎頻譜障礙(AQP4-IgG-NMOSD),不符合MS或其他已知神經炎性疾病的典型標準,最常見的是視神經炎(ON),其次是脊髓炎,急性播散性腦脊髓炎(ADEM)或ADEM樣呈現(例如,腦干攻擊),其特征是神經功能障礙的單相或復發性過程[2,4,5]。
據以往研究,MOG抗原可能是在血-腦屏障的通透性增加時(如CNS感染時)滲漏入外周而被外周免疫系統識別,通過腸道菌群輔助MOG特異性地CD4+T細胞和B細胞生成和活化,促進了MOG抗體在外周淋巴器官中產生。MOG抗體為炎性脫髓鞘疾病的致病性抗體,血腦屏障的功能再次遭受破壞后,MOG-IgG通過被動擴散或通過滲漏的血腦屏障到達CSF中[6]。MOG-Ab與T細胞協同作用后具有同致病能力,被動擴散到達CSF后通過細胞毒作用,誘導補體和抗體介導神經系統損傷[7]。血清MOG-Ab的檢測被視為確診MOG-AD的金標準,然而當血清抗體陰性、臨床高度懷疑為NMOSD、MOG、AQP4-Ab病例時,行腦脊液MOG-Ab檢測可以為診斷提供幫助。臨床上仍有極少數僅在腦脊液中存在MOG-Ab的MOG-AD患者,這類患者的致病抗體誘導產生的B細胞可以僅存在于中樞神經系統中,反映了MOG-Ab在鞘內合成的可能而非血清中的抗體被動擴散至中樞神經系統中[8]。
神經免疫的相互作用可導致許多神經系統疾病的發生,雖然中樞神經系統免疫監視的機制尚不明確,但可能是導致該病發生的重要因素。硬腦膜中存在穩定的白細胞群體,對參與適應性免疫功能的免疫群體進行整體免疫組化研究后發現,T細胞和組織相容性復合體Ⅱ(MHCⅡ)+抗原呈遞細胞(APCs)并非均勻分布在整個硬腦膜組織中,而是高度分布在硬腦膜竇周圍(鼻竇),成為免疫樞紐。動物實驗證明,在使用小劑量示蹤劑注射于小鼠紋狀體實質內后,最終發現許多維持大腦穩態的蛋白質積累于硬腦膜周圍。硬腦膜脈管系統可以進行平穩持續的T細胞監視,硬腦膜竇相關的APCs可以捕獲腦脊液抗原,并且硬腦膜竇是循環的自我反應性T細胞遇到其同源抗原的關鍵部位,包括EAE/MS中的MOG反應性T細胞。在研究自身免疫性腦脊髓炎(EAE)中 CNS 抗原的局部呈遞與自身免疫性神經炎癥的相關性時,在EAE發病后,MHCⅡ+APC 數量沒有顯著變化,但在硬腦膜內積累了大量的T細胞,通過液相色譜-質譜(liquid chromatography-mass spectrometry,LC-MS)分析顯示硬腦膜存在 MOG 積累。同時,在實驗中也觀察到了在EAE發病期間MOG 反應性T細胞在外周的激活、在硬腦膜中的積累。在EAE急性發作時,所有CNS脈管系統都被炎性浸潤,但T細胞可以從不同部位浸潤CNS組織,從而導致不同部位的神經系統疾病[4]。以上機制為MOG-AD存在原發于中樞神經系統的情況提供了有力依據。
放射學和血清學確診的MOGAD的病理學特征是靜脈和融合性白質脫髓鞘。這表明MOG-AD脫髓鞘的方式介于MS和具有靜脈型的ADEM之間。炎性浸潤主要由CD4+T細胞和粒細胞組成,而CD8+T細胞占主導地位。皮質內的脫髓鞘病變更為常見。在活動性病變中可以觀察到補體沉積,但在星形膠質細胞或神經膠質母細胞上未觀察到補體沉積,因為MOGAD不是星形細胞病變,使得AQP4未受到損傷,由此可以很容易地與AQP4陽性的NMOSD和MS區分開[3]。對1例僅在CSF中具有MOG-Abs的患者的大腦,脊髓、神經根和軟腦膜的尸檢后發現,該患者存在大量的組織破壞和腦橋和脊髓的脫髓鞘,其主要特征是相對稀疏的軸突、靜脈、皮下及皮質匯合的原發性脫髓鞘,反應性神經膠質增生和主要由粒細胞,巨噬細胞和CD4+T細胞構成的炎性浸潤,并在炎性浸潤物中,發現了IgG沉積物,證實了僅在CSF中具有MOG-Abs的炎性浸潤相關病變確實存在[7]。
雖然該患者中樞神經系統中MOG-Ab的滴度較低,但仍考慮致病性抗原滴度較低也可導致脊髓炎的發生[8~10]。一項對100例髓鞘少突膠質細胞糖蛋白(MOG)抗體患者的腦脊液163次腰椎穿刺的結果研究發現:在少數有鞘內IgG合成的樣本中,僅在急性疾病發作期間發現鞘內MOG-IgG,3/4隨訪的患者CFS中MOG-IgG僅短暫存在。3/6 寡克隆帶(OCB)陽性患者中的OCB只是短暫出現一次。此研究說明了對多數MOG-IgG陽性EM患者的CSF進行定性和定量研究后,提示如果鞘內IgG合成存在,合成率往往較低,且主要在急性發作時短暫存在[11]。
據報道,在急性期靜脈注射甲潑尼龍(IVMP)可有效治療MOG抗體陽性ON和(或)脊髓炎(劑量為每天1~2 g,連續3~5 d),患者癥狀可能得到緩解[2],較嚴重或類固醇治療無效的處于疾病急性期的患者應早期進行血漿置換和靜脈內免疫球蛋白(IVIG)治療[3]。該患者急性期經激素沖擊療法治療12 d后,復查CSF中MOG-Ab由弱陽性轉為陰性,從患者癥狀以及輔助檢查都可以體現出急性期甲潑尼龍沖擊療法治療有效。
對于長期復發的預防性治療,潑尼松龍的長期治療(體重>40 kg的患者,劑量>10 mg/d,體重≤40 kg的患者,劑量>5 mg),靜脈內免疫球蛋白(誘導療程劑量為2 g/kg,隨后為每月) 1 g/kg輸注,利妥昔單抗、霉酚酸酯、氨甲蝶呤或硫唑嘌呤都有報道,以減少MOG-AD復發。但是,利妥昔單抗治療可能與首次輸注后幾周內的新發作有關,可能是由于B細胞活化因子和自身抗體水平的暫時升高,正如一些AQP4陽性NMO患者所觀察到的。那他珠單抗治療不能有效地預防復發,而奧法木單抗(ofatumumab)的治療卻能將年復發率從2.1降低至0.66。一項研究發現,與非類固醇維持療法(38%)相比,接受類固醇維持療法的患者治療失敗率更低。口服類固醇作為免疫抑制劑輔助治療的作用尚不清楚。一項研究發現,接受硫唑嘌呤治療但未輔以口服類固醇治療的MOG抗體ON和(或)脊髓炎患者比接受聯合治療的患者更容易復發。此外,復發似乎更頻繁地發生時口服潑尼松龍的劑量下降到低于10 mg/d或停止類固醇。干擾素β治療已被證明是無效的,并且可增加患病的風險。同樣,醋酸格拉替雷在兒童患者中也被證明無效[2,3]。因此,該患者靜脈滴注12 d甲潑尼龍治療后,改為口服甲潑尼龍片,起始量為60 mg/d,每15 d減4 mg至停藥,患者現未出現復發。但尚未發現高度特異性的放射學表現來區分MOGAD和非MOG抗體病例。
3 結 論
該患者僅腦脊液中MOG抗體為陽性,考慮神經免疫反應可能于中樞神經系統的硬腦膜竇中完成,相關抗體未滲入外周。因此,臨床上考慮為中樞神經系統炎性脫髓鞘疾病的患者,血清中相關抗體均為陰性時,要注意完善腦脊液中相關抗體檢測,以避免漏診及誤診。
