土壤鐵氧化物–亞鐵的相互作用及其環境影響研究進展①
2023-09-22 03:11:00余光輝
土壤 2023年4期
關鍵詞:研究
姚 遠,余光輝,滕 輝*
土壤鐵氧化物–亞鐵的相互作用及其環境影響研究進展①
姚 遠1,2,余光輝1,滕 輝1*
(1 天津大學地球系統科學學院,表層地球系統科學研究院,天津 300072;2 中國地質調查局西安地質調查中心,西安 710119)
鐵氧化物和溶液相亞鐵常在厭氧土壤環境中共存。鐵氧化物能夠加快亞鐵的氧化速率,且控制亞鐵氧化成礦產物的類型,同時,亞鐵與鐵氧化物組成的系統是一種良好的還原劑,能夠有效還原重金屬及降解有機污染物。另一方面,亞鐵能夠催化鐵氧化物晶相轉變,導致鐵氧化物結構和表面性質發生改變,進而影響相關重金屬、有機質的環境行為。本文綜述了鐵氧化物催化亞鐵氧化成礦、鐵氧化物–亞鐵系統還原污染物以及亞鐵催化鐵氧化物相變的反應機制及影響因素,最后,對未來在自然土壤中研究鐵氧化物–亞鐵界面反應及其環境影響進行了展望。
鐵氧化物;亞鐵氧化;礦物相變;有機污染物;重金屬
鐵元素(Fe)是地殼中含量第四的元素,與其前后的Al和Ca金屬元素不同, Fe元素具有更強的化學反應性,廣泛參與環境中各類氧化還原反應,Fe的電子排布為[Ar]3d64s2,而[Ar]3d64s0和[Ar]3d54s0結構的穩定性確保Fe能以Fe(Ⅱ)和Fe(Ⅲ)的形式存在,同時,溶液相Fe3+轉化為Fe2+的標準還原電位為0.771 V,這使得這類反應在自然條件下普遍發生。由于Fe元素含量豐富以及良好的氧化還原反應活性,Fe(Ⅱ) 的氧化與Fe(Ⅲ) 的還原驅動著環境眾多元素的轉化與物質循環,因此,研究Fe元素在環境中的氧化還原反應具有重要研究意義。
自然環境中,Fe主要以Fe(Ⅱ) 和Fe(Ⅲ) 形式存在,鐵(氫)氧化物(文中統稱為鐵氧化物)是土壤中最活躍的組成成分之一[1],由于其良好的吸附能力及生物反應性,鐵氧化物對土壤中重金屬的鈍化與固定、有機質的礦化與積累、養分元素的利用等眾多環境過程有著重要影響[2]。Fe(Ⅱ) 相比Fe(Ⅲ) 更易溶解,導致Fe(Ⅱ) 在生物可利用的形式中豐度較高,Fe(Ⅱ) 可以通過巖石的物理、化學風化作用進入到環境中,也可以通過厭氧環境下化學還原劑(如硫化物)或異化鐵還原菌還原Fe(Ⅲ) 礦物產生。這也使得溶液相Fe(Ⅱ) 常與鐵氧化物共存,近二十年來,隨著人們對Fe的氧化還原反應的研究,發現相比溶液相Fe(Ⅱ),與鐵氧化物表面結合Fe(Ⅱ) 具有更強的還原反應性,例如:早在1999年Buerge和Hug[3]發現Fe(Ⅱ) 與鐵氧化物組成的系統能夠快速還原重金屬Cr(Ⅵ)。之后圍繞這一思路,一系列研究探索了厭氧體系中,Fe(Ⅱ) 與鐵氧化物系統在還原降解眾多重金屬及有機污染物的反應,包括Cr(Ⅵ)、Hg(Ⅱ)、As(Ⅴ) 等重金屬污染物[4]以及四氯化碳、硝基芳香烴、亞硝酸鹽等有機污染物[5-10];并詳細研究了這一系統反應性的影響因素,這類反應在土壤原位修復領域具有良好的應用前景。同時,在上述反應中,研究還發現鐵氧化物表面具有模板效應,能夠控制Fe(Ⅱ) 氧化成礦產物的類型。如Larese-Casanova等[11]發現反應中存在的針鐵礦誘導Fe(Ⅱ) 氧化形成針鐵礦,后續的研究表明在有氧環境下,這種模板效應依然存在,同時也發現鐵氧化物的存在能夠顯著加快Fe(Ⅱ) 的氧化速率。另一方面,研究發現溶液相亞鐵能夠催化弱結晶態鐵氧化物發生晶相轉化形成結晶度高的鐵氧化物。得益于斯堡爾光譜技術的應用,Williams 和Scherer[12]首先采用同位素標記及穆斯堡爾光譜的方法證實了Fe(Ⅱ) 與鐵氧化物之間的電子傳遞作用,后續眾多研究也報道了Fe(Ⅱ) 與鐵氧化物之間的電子傳遞和原子交換現象[13-14]。Scherer等人的一系列研究表明Fe(Ⅱ)催化鐵氧化物的晶體相變反應是由Fe(Ⅱ) 與鐵氧化物之間的電子傳遞/原子交換作用驅動的[15]。基于眾多界面電子傳遞和原子交換的研究,大多數學者認為亞鐵催化鐵氧化物相變的實質是電子傳遞和原子交換作用導致的鐵氧化物固相轉化或溶解重結晶,但目前關于鐵氧化物如何發生溶解以及礦物如何成核生長仍缺乏直接證據,仍需持續研究關注。同時,在這一過程中,由于鐵氧化物的礦物學的轉變會導致其表面性質與反應活性的改變,進而影響與之共存的重金屬、有機質等物質的環境行為,具有重要環境意義。
目前,鐵氧化物–亞鐵之間的相互作用機制及其環境影響已在室內控制實驗中取得較好的進展,但實際土壤環境復雜多變,鐵氧化物–亞鐵之間的相互作用常處于動態變化之中,使得研究自然條件下鐵氧化物–亞鐵之間的界面反應變得更為困難,也導致預測與之相關的重金屬、有機物等物質的環境行為面臨十足的挑戰。本文就鐵氧化物–亞鐵之間的相互作用以及相關環境影響的研究進行了總結,并以此為基礎提出相關研究今后的重點和方向,進一步加快相關研究的發展和應用。
1 土壤常見鐵氧化物類型及現代分析技術
鐵氧化物普遍分布于各種類型的土壤中,由于表面反應活性高,在自然環境中,很難找到一個沒有鐵氧化物參與的反應[16]。目前,已報道的鐵氧化物共16種[17],其中土壤中最常見的有水鐵礦、纖鐵礦、針鐵礦、赤鐵礦等。水鐵礦(Fe(OH)3),經常分布在灰壤中的溫和或冰冷的區域、酸性礦山廢水污染區域及淹水水稻土中,具有比表面積大、結晶度差、表面反應活性高等特點,通常難以在環境中穩定持續存在,具有較高的標準吉布斯自由能((–469.9±1.2) kJ/mol)[18],易于轉變為結構更穩定的鐵礦物。常見的相變產物有磁鐵礦、針鐵礦、赤鐵礦,因此水鐵礦是一些熱力學穩定鐵氧化物的前驅體。此外,由于水鐵礦粒徑較小,且晶體結構復雜,目前關于水鐵礦的結構尚未有統一的認識。纖鐵礦(γ-FeOOH)常見于季節性交替氧化還原波動的土壤環境中,是一種亞穩態礦物,可以作為其他鐵氧化物的前驅礦物[19],一般易于轉化為針鐵礦。纖鐵礦結構為陰離子的立方密堆積(ccp)的伯姆石層狀結構,屬斜方晶系。針鐵礦(α-FeOOH)幾乎存在于所有類型的土壤、湖泊及河流沉積物中[20],通常是由黃鐵礦或磁鐵礦等其他鐵礦物風化而來,屬于斜方晶系,結構為立方體緊密堆積結構(ccp),一般與纖鐵礦為同質多象,是一種熱力學穩定性較高的鐵氧化物,不易轉變為其他鐵氧化物。赤鐵礦(Fe2O3) 普遍分布于熱帶、亞熱帶溫度較高的土壤和沉積物中[17, 21]。赤鐵礦具有六方陰離子緊密堆積(hcp)的剛玉結構,熱力學穩定性強,通常不會轉變為其他鐵礦物相。在一定條件下,上述各類型鐵氧化物晶體之間可以互相轉化,在大多數自然條件下,由于吉布斯自由能差產生的驅動力,鐵氧化物趨向形成熱力學穩定的礦物(如針鐵礦與赤鐵礦),相關熱力學參數見表1。
自然土壤環境多樣進而導致土壤鐵氧化物的化學結構與組成復雜,目前鐵氧化物的礦物特征已經能夠得到很好的表征認識(表2)。常規的傳統表征技術手段如X射線粉晶衍射分析(XRD)、掃描電子顯微鏡(SEM)、透射掃描電子顯微鏡(TEM)、傅里葉變換紅外光譜(FTIR)、X 射線光電子能譜(XPS)、動態光散射(DLS)、靜態光散射(SLS)等能夠較好地表征鐵氧化物基本的組成、形貌、粒徑、官能團以及分散與凝聚狀態等特征,但由于上述技術的局限性,無法對礦物相關的晶體結構、價態、分子/原子的排列方式等特征進行更加深入的認識,限制了人們對鐵氧化物及相關界面反應機理的探索。目前得益于穆斯堡爾譜(M?ssbauer spectra)、原子力顯微鏡(AFM)、同步輻射X射線吸收光譜(XAS)、球差校正掃描透射電子顯微鏡(Cs-STEM)等一系列現代分析技術的快速發展,為人們了解鐵氧化物及其相關界面反應提供了強大支持[24]。
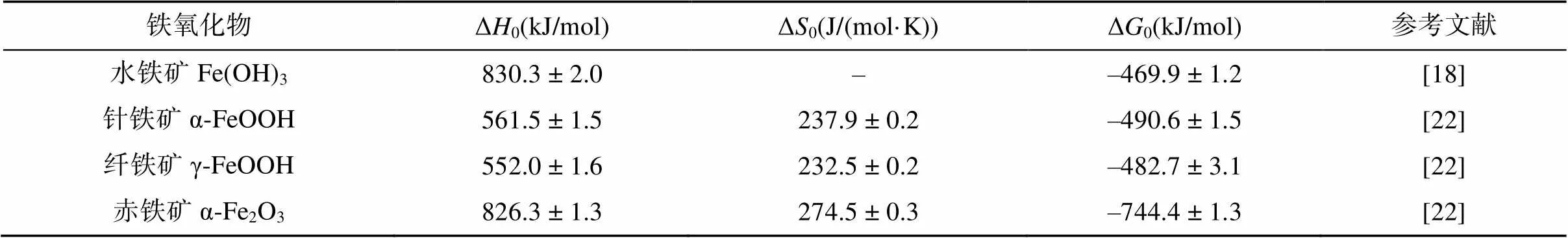
表1 土壤常見鐵氧化物的熱力學參數
注:焓(Δ)和吉布斯自由能(Δ)的參數條件為298 K、1 bar。

表2 研究礦物的現代分析技術匯總[23]
2 鐵氧化物催化Fe(Ⅱ) 氧化成礦
Fe(Ⅱ) 的氧化包括非生物(化學)和生物過程[25],主要受控于自然環境中pH和O2濃度[26-27]。Fe(Ⅱ) 的非生物氧化成礦是土壤中次生鐵氧化物形成的重要來源。一方面,亞鐵的氧化可以通過溶液中的均相反應(homogeneous reactions)進行;另一方面,由于環境中存在各種礦物,因此亞鐵的氧化經常在礦物影響下,以非均相反應(heterogeneous reactions)的方式進行。由于鐵氧化物在土壤中大量存在,且反應活性高,因此許多研究關注在鐵氧化物存在下,Fe(Ⅱ) 的氧化成礦過程。目前,眾多研究發現鐵氧化物表面可以催化Fe(Ⅱ) 氧化成礦,提高Fe(Ⅱ) 氧化速率,且明顯控制新生鐵氧化物類型與特征。
眾多鐵氧化物具有半導體的性質,能夠促進礦物的電子傳遞過程[14],從而加快Fe(Ⅱ) 的氧化速率。同時,研究發現Fe(Ⅱ) 的氧化速率與鐵氧化物結晶度以及吸附能力密切相關。Tamura 等人[28]研究了中性pH條件下,水鐵礦、針鐵礦和纖鐵礦對Fe(Ⅱ) 氧化動力學的影響,發現在水鐵礦存在時,Fe(Ⅱ) 的氧化速率大概是針鐵礦、纖鐵礦存在時的3倍,因此提出鐵氧化物表面催化Fe(Ⅱ) 氧化的速率與其結晶度直接相關。而Jones等[29]研究發現在酸性條件下(pH 4.5 ~ 5.5),鐵氧化物依然能顯著加快Fe(Ⅱ) 的氧化速率,且Fe(Ⅱ) 的氧化速率正比于鐵氧化物表面吸附態Fe(Ⅱ) 的濃度,由此認為鐵氧化物催化Fe(Ⅱ) 氧化的速率是由鐵氧化物對Fe(Ⅱ) 的吸附量所控制;類似地,Park和Dempsey[27]也報道了中性低氧條件下,水鐵礦催化Fe(Ⅱ) 氧化速率受控于吸附態Fe(Ⅱ)的含量。然而,上述研究中反應體系限于純反應系統,沒有考慮自然土壤系統的多相性。近期Chen 和Thompson[30]研究了自然土壤環境下鐵氧化物對Fe(Ⅱ) 的氧化成礦的影響,通過去除自然土壤中鐵氧化物,與原土壤進行對比試驗,發現土壤中鐵氧化物的存在同樣會加快Fe(Ⅱ) 的氧化速率。
此外,鐵氧化物能夠作為Fe(Ⅱ) 氧化成礦的模板,影響成礦產物的類型。通常,Fe(Ⅱ) 在溶液中的均相氧化會形成無定形Fe(Ⅲ)-OH絡合物或亞穩態鐵礦物(如水鐵礦或纖鐵礦)[31],而在結晶態鐵氧化物存在下,Fe(Ⅱ)氧化會形成熱力學穩定的鐵氧化物(如針鐵礦或赤鐵礦)[6,11,31]。在這類反應中,Fe(Ⅱ) 在鐵氧化物表面的反復吸附和氧化促使新的鐵氧化物相的積累,導致新生鐵氧化物相通常類似于吸附的鐵氧化物表面。如:在厭氧體系中,Chun等人[6]在研究Fe(Ⅱ)-針鐵礦系統還原降解4–氯硝基苯和三氯硝基甲烷的試驗中,發現在針鐵礦作用下,Fe(Ⅱ) 氧化后新形成的礦物也為針鐵礦;同樣,Larese-Casanova等人[11]發現針鐵礦誘導Fe(Ⅱ) 形成針鐵礦,而在磁鐵礦與磁赤鐵礦表面促使Fe(Ⅱ) 氧化形成纖鐵礦。另一方面,在有氧環境下,研究者發現針鐵礦表面同樣促使Fe(Ⅱ) 氧化形成結晶度高的針鐵礦[31]。此外,Chen和Thompson[30]發現在復雜的自然土壤環境中,上述鐵氧化物的模板效應依然存在,即Fe(Ⅱ) 氧化產物類型強烈受控于土壤中預先存在的鐵氧化物,由此可見鐵氧化物對Fe(Ⅱ) 氧化產物類型的控制作用在自然環境中普遍發生。此項研究將先前類似的研究擴展至復雜的自然土壤環境,后續研究可在此基礎上繼續探索自然土壤環境中鐵氧化物對Fe(Ⅱ) 的氧化成礦的影響。同時,當前的研究僅限于純的鐵氧化物反應體系,在土壤中,鐵氧化物在形成過程中晶格常摻雜各種離子或分子雜質或者發生陽離子同晶替代,這些現象都會改變原來鐵氧化物的結晶度、形貌、比表面積等礦物學特征以及表面反應活性。這些性質的改變是否會改變鐵氧化物催化Fe(Ⅱ) 氧化的反應動力學及其對產物類型的控制,值得后續深入研究。
3 鐵氧化物–Fe(Ⅱ)還原污染物及其反應性的影響因素
3.1 鐵氧化物–Fe(Ⅱ)還原污染物
目前眾多研究報道了鐵氧化物與溶液相亞鐵組成的系統還原降解眾多污染物,包括四氯化碳、硝基芳香烴、亞硝酸鹽等污染物[5-10]以及Cr(Ⅵ)、Hg(Ⅱ)、As(Ⅴ) 等重金屬污染物[3-4](表3)。在這些還原污染物的反應中,無論使用何種類型的鐵氧化物,相同污染物的反應產物都是相似的。其中有機污染物中的脫鹵反應普遍發生,例如四氯化碳(CCl4)脫氯后生成氯仿[5, 32]。
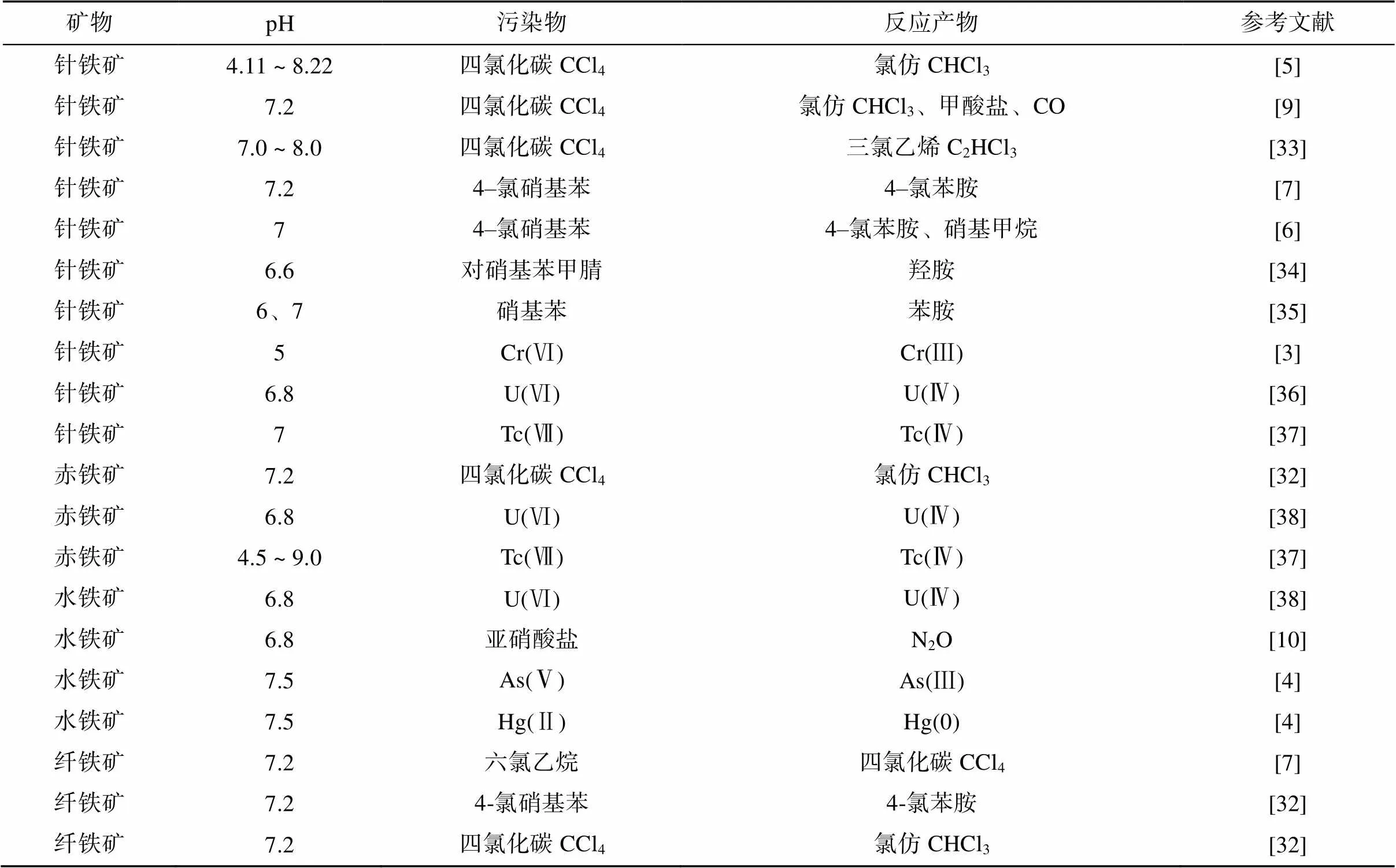
表3 鐵氧化物–Fe(Ⅱ)系統還原污染物的相關報道

當前關于鐵氧化物與亞鐵組成的系統還原降解污染物的研究尚局限于單一的反應過程,缺少環境中多組分參與耦合反應的研究,后續研究需要考慮其他可能參與反應的物質,以模擬實際土壤中該類反應的發生。此外,在一些受污染土壤中存在有機無機復合污染的情況,該方面的研究目前較為薄弱,未來值得進一步關注。
3.2 鐵氧化物–Fe(Ⅱ) 還原反應性的影響因素
在鐵氧化物–Fe(Ⅱ)系統還原降解污染物的研究中,即使相同類型的鐵氧化物和相同的污染物,吸附在鐵氧化物表面上的Fe(Ⅱ) 的還原反應性也存在較大差異。該還原反應性與鐵氧化物的聚集狀態、暴露晶面等礦物性質以及反應pH、Fe(Ⅱ) 濃度、有機質的存在等反應條件密切相關。
鐵氧化物顆粒的聚集會導致吸附在其表面的Fe(Ⅱ) 的還原反應性降低。例如,Amonette等人[5]研究發現,針鐵礦顆粒的聚集導致其表面可用位點減少從而使得四氯化碳的降解速率降低。常用的有機緩沖液,如 MOP、HEPES等可增強鐵氧化物納米顆粒的聚集狀態并降低這些系統的還原反應性[47]。另一方面,相比大顆粒鐵氧化物,粒徑較小的鐵氧化物由于更大反應比表面積以及更高的表面反應能,通常具有更強的氧化還原反應性。然而,Cwiertny等人[48]在研究納米針鐵礦與亞鐵組成的系統還原硝基苯的反應中,發現不同粒徑的納米針鐵礦都發生顯著的聚集,形成微米級的密集聚集體,當還原速率以比表面積歸一化后,卻發現粒徑小的針鐵礦納米顆粒系統還原反應性低于較大粒徑針鐵礦的還原反應性。因此,直接使用比表面積來評估聚集顆粒懸浮液中表面反應性的差異并不合理,為了真實地評估這類反應中的納米級尺寸效應,需要開發新的方法來量化濕納米顆粒在懸浮液中可用于吸附和反應的表面積量。
鐵氧化物的暴露晶面能夠影響吸附態Fe(Ⅱ)的還原反應性。Chun 等人[6]研究發現針鐵礦{021}晶面損失,會降低Fe(Ⅱ)–針鐵礦系統對4–氯硝基苯和三氯硝基甲烷還原反應性;Huang等人[21]研究表明,赤鐵礦{110}晶面可以將Fe(Ⅱ) 限制在五配位排列中,而{001}晶面可以將Fe(Ⅱ) 限制為六配位排列,相比在赤鐵礦{001}面上的六配位排列,赤鐵礦{110}面上的五配位排列表現出更強的Fe(Ⅱ)吸附能力。更重要的是,赤鐵礦{110}晶面上Fe(Ⅱ)的五配位排列方式比赤鐵礦{001}面上的六配位排列能更加有效地催化過氧化氫轉化為羥基自由基,從而降解各種有機污染物。因此,鐵氧化物的晶面特異反應性對于環境修復具有廣泛的意義。目前部分研究聚焦開發具有優勢晶面的鐵氧化物顆粒,以提高 Fe(Ⅱ)–鐵氧化物系統還原降解重金屬及有機污染物的能力,未來該方面的研究值得持續關注。
pH通過影響Fe(Ⅱ) 在鐵氧化物表面的吸附行為,從而影響Fe(Ⅱ)–鐵氧化物系統的還原反應性。自然環境中pH范圍一般為5 ~ 7.5,當 pH<5時,礦物表面吸附的Fe(Ⅱ) 的量非常有限,導致還原反應性低;而pH>7.5時,則會形成Fe(Ⅱ) 沉淀物。在自然pH范圍內,隨著 pH 的增加,礦物對Fe(Ⅱ) 的吸附變強,從而提高還原反應性。另一方面,pH會影響吸附于礦物表面的Fe(Ⅱ) 類型。Zhang等人[49]研究了Fe(Ⅱ) 在纖鐵礦上的吸附,發現在纖鐵礦表面形成兩類Fe(Ⅱ) 表面絡合物,其中,Fe(Ⅱ)單羥基表面配合物 (≡FeⅢOFeⅡOH-)是主要類型,而(≡FeⅢOFeⅡ)+是次要類型。后來研究采用表面絡合模型(surface complexation model) 擬合Fe(Ⅱ) 在氧化鐵上的吸附的研究[50](圖1),方程(1)和(2)顯示了這兩種穩定的表面Fe(Ⅱ)絡合物的形成。
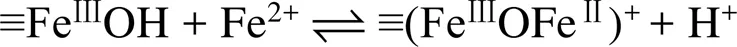

這兩種形式的Fe(Ⅱ) 依賴于溶液pH,且研究表明4–氯硝基苯、U(Ⅵ) 還原速率與以(≡FeⅢOFeⅡOH-)形式存在的吸附態Fe(Ⅱ) 濃度呈正比關系[50-51]。然而,隨后的研究發現吸附態Fe(Ⅱ) 與鐵氧化物結構Fe(Ⅲ) 發生電子傳遞作用,尤其在低Fe(Ⅱ) 濃度條件下,吸附態Fe(Ⅱ) 的存在是瞬時的,Fe(Ⅱ) 會迅速與鐵氧化物結構Fe(Ⅲ)發生界面電子傳遞并產生新的鐵氧化物[12, 43],說明表面絡合模型中所提到的Fe(Ⅱ) 表面絡合物并不會在反應中穩定存在,因此,該模型在這類反應中的適用性需進一步研究討論。
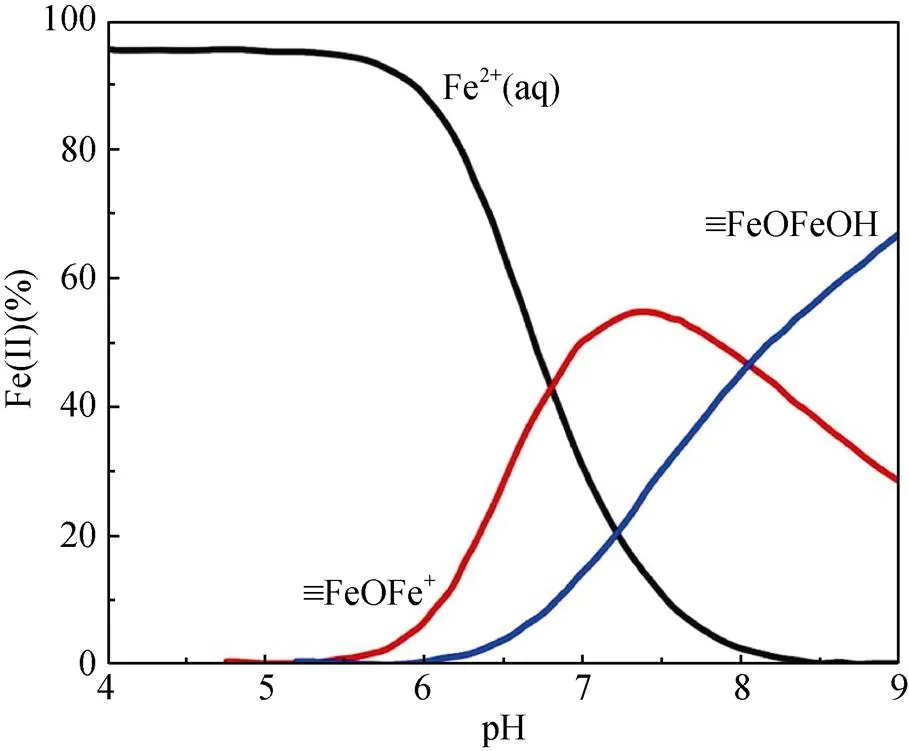
圖1 吸附于赤鐵礦表面的Fe(Ⅱ) 類型的分布[24]
溶液相Fe(Ⅱ) 的濃度也是影響礦物表面Fe(Ⅱ) 還原反應性的重要因素。在低Fe(Ⅱ) 濃度下,Fe(Ⅱ) 濃度的增加會提升鐵氧化物對Fe(Ⅱ) 的吸附量,這促進了水溶液 Fe(Ⅱ) 和晶格Fe(Ⅲ) 之間的界面電子傳遞,并產生更快的還原反應。在高Fe(Ⅱ) 濃度下,尤其是吸附的 Fe(Ⅱ) 量超過了鐵氧化物表面單層吸附位覆蓋量,導致界面電子傳遞速率快速降低,因此氧化還原反應性受到抑制[15]。
有機質(如富里酸、胡敏酸、胡敏素等)可以通過靜電作用、氫鍵作用、配體交換和疏水作用吸附在鐵氧化物表面[52],從而影響吸附態Fe(Ⅱ) 的反應性。Colón等人[34]研究表明有機質主要通過絡合吸附于針鐵礦表面的Fe(Ⅱ),抑制吸附態Fe(Ⅱ) 與污染物之間的電子傳遞作用,從而降低Fe(Ⅱ)–針鐵礦體系的還原反應性。同樣地,Vindedahl等人[53]也發現一系列有機質的添加,包括Pahokee Peat 腐植酸、Elliot土壤腐植酸、Suwannee River腐植酸、Suwannee River NOM、Suwannee River富里酸I、Suwannee River富里酸Ⅱ和Pahokee Peat 富里酸,都在不同程度上抑制了Fe(Ⅱ)–針鐵礦系統對4–氯硝基苯 (4-ClNB) 的還原反應性。同時在這些NOM-Fe(Ⅱ)–針鐵礦體系中,發現隨著有機質的分子量和氮、碳和芳烴含量的增加,4-ClNB的還原降解速率更快,而隨著羧基、氧、雜脂族和脂肪族含量的增加,還原速率更慢,這一發現為預測有機質與Fe(Ⅱ)、鐵氧化物組成的系統的還原反應性提供了依據。
4 亞鐵催化鐵氧化物相變及其環境影響
環境中,各類型鐵氧化物晶體之間可以互相轉化,鐵氧化物之間的晶相轉化有溶解再結晶或直接固相轉化兩種方式。溶解再結晶是指弱結晶的鐵氧化物先溶解,產生Fe(OH)2+和 Fe(OH)+等羥基鐵離子,然后再通過聚合作用形成結晶度高的鐵氧化物;固相直接轉化機制是弱結晶鐵氧化物通過脫水和結構重排直接轉化為結晶度高的鐵氧化物[54]。在有氧環境中,針鐵礦和赤鐵礦是兩種熱力學穩定的鐵氧化物相,也是大多數鐵氧化物轉化的最終礦物相。土壤環境中,結晶度差的水鐵礦一般作為晶體鐵氧化物的前驅礦物,由于比表面積大及表面活性高,水鐵礦對重金屬等污染物具有很高吸附容量,因此研究水鐵礦的相變具有重要環境意義,目前,亞鐵催化水鐵礦相變受到廣泛關注。
4.1 亞鐵催化鐵礦物相變的反應機制
亞鐵催化鐵氧化物相變的實質是Fe(Ⅱ) 與鐵氧化物之間的界面電子傳遞和原子交換作用,使得鐵氧化物直接發生固相轉化或溶解重結晶[15]。Williams 和Scherer[12]首先采用同位素標記及穆斯堡爾譜的方法證實了這一現象。這種界面電子傳遞作用與鐵氧化物結構中一些晶格Fe(Ⅲ) 的還原溶解行為密切相關[14]。對此Handler等人[13]提出了Fe(Ⅱ) 與鐵氧化物之間電子傳遞的模型,很好地揭示了Fe(Ⅱ) 與鐵氧化物之間電子傳遞作用的具體反應過程 (圖2)。近年來,Scherer課題組系統研究了溶液相Fe(Ⅱ) 催化各類型鐵氧化物的晶體相變過程,結果也表明:鐵氧化物結構態Fe(Ⅲ) 與Fe(Ⅱ) 發生原子交換,Fe(Ⅱ) 進入鐵氧化物表面或結構中被氧化為Fe(Ⅲ),形成次生鐵氧化物,而原始鐵氧化物中結構態Fe(Ⅲ) 則被還原,發生溶解釋放到溶液中去,重新成核結晶形成新的更加穩定的鐵氧化物[15]。而Jones等人[55]研究發現吸附于鐵氧化物表面的硅酸鹽能夠抑制Fe(Ⅱ) 的吸附,導致Fe(Ⅱ) 與礦物結構態Fe(Ⅲ) 之間的原子交換減少,從而抑制了Fe(Ⅱ) 催化水鐵礦晶相轉化過程;此外,還發現水鐵礦晶格內的硅酸鹽并沒有降低Fe(Ⅱ) 與水鐵礦之間的原子交換,但依然抑制了水鐵礦的相變。這表明水鐵礦的溶解過程釋放出來的硅酸鹽抑制了新礦物的結晶,由此也進一步支持了Fe(Ⅱ) 催化的Fe(Ⅲ) 礦物相變的溶解重結晶機制。
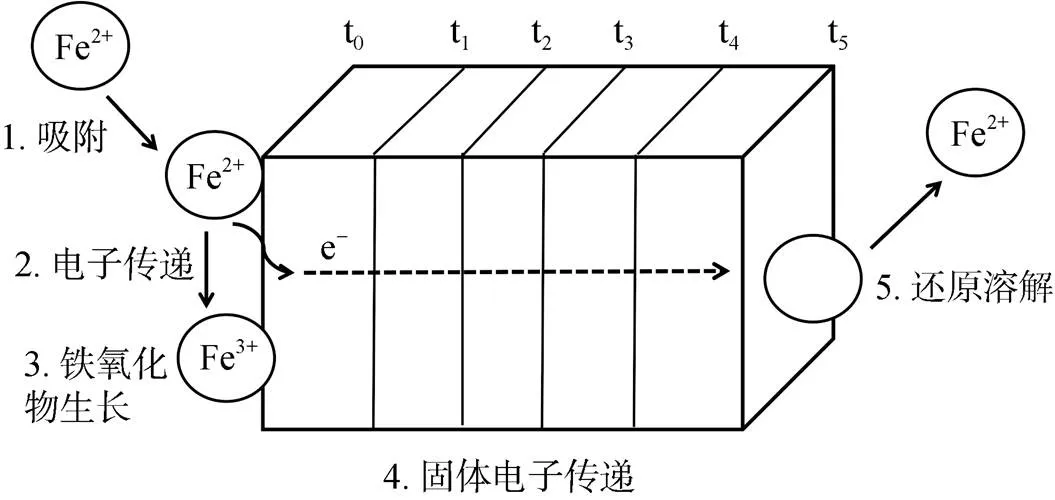
圖2 亞鐵與鐵氧化物之間的電子傳遞機制模型(修改自Handler等[13])
此外,Boland等人[56]提出一種新的Fe(Ⅱ) 催化水鐵礦相變過程的解釋(圖3):Fe(Ⅱ) 吸附到水鐵礦表面后通過電子傳遞作用立即氧化成 Fe(Ⅲ),由此產生的電子傳遞到水鐵礦晶格Fe(Ⅲ),將Fe(Ⅲ) 還原為Fe(Ⅱ) 釋放到溶液中,氧化產生的Fe(Ⅲ) 會沉淀為活性水鐵礦,其作為新礦物生長結晶的核,同時也能進一步吸收溶液中的Fe(Ⅱ),這種作用不斷重復進行,直至活性水鐵礦相變為針鐵礦。目前,一系列鐵氧化物與Fe(Ⅱ) 之間的電子傳遞及原子交換過程的發現,極大促進了人們對亞鐵催化水鐵礦相變的過程認識,但除了動力學以及晶格離子溶出現象,尚缺乏直接的實驗證明水鐵礦的實際晶相轉變過程,未來仍需進一步研究。
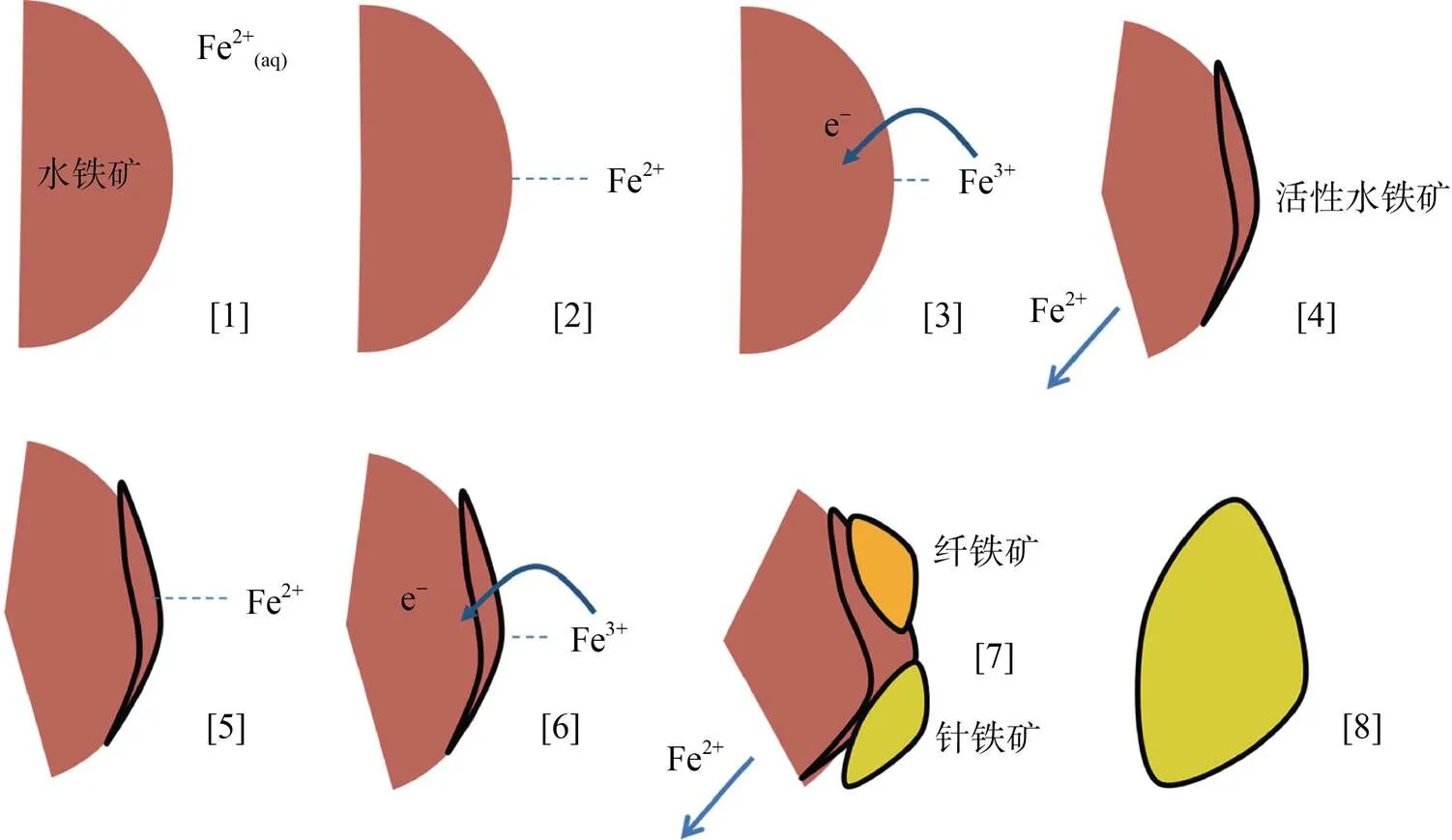
圖3 Boland等提出的水鐵礦轉變機理模型(修改自Boland等[56])
4.2 亞鐵催化鐵氧化物相變的影響因素
Fe(Ⅱ) 催化鐵氧化物相變受水溶液pH及Fe(Ⅱ) 濃度影響。其中,pH通過影響Fe(Ⅱ) 在礦物表面的吸附行為來影響Fe(Ⅱ) 與礦物之間的電子傳遞過程,從而控制鐵氧化物的晶相轉化過程。Fe(Ⅱ) 在鐵氧化物表面的吸附會產生H+,隨著溶液pH的升高,H+濃度降低,越有利于Fe(Ⅱ) 的吸附,從而加快鐵氧化物的相變速率。另一方面,溶液pH也會控制鐵氧化物相變的礦物類型,Boland等人[56]研究了一定pH范圍內(6.17 ~ 7.26),Fe(Ⅱ)催化水鐵礦相變反應,發現水鐵礦相變的中間產物纖鐵礦相通常在較低的 pH條件下普遍存在,較高pH通常會導致相變的最終產物針鐵礦的快速形成。而在Hancel等人[57]的研究中,發現pH=8時會促使水鐵礦完全轉化為磁鐵礦沉淀。另一方面,溶液Fe(Ⅱ) 的濃度也能影響鐵氧化物相變產物類型。研究表明在低濃度Fe(Ⅱ) (<1.0 mmol/g水鐵礦)條件下,水鐵礦通過溶解和再沉淀轉化為纖鐵礦和針鐵礦;然而,在較高濃度Fe(Ⅱ) (>1.0 mmol/g水鐵礦)條件下,則有利于磁鐵礦的沉淀的形成[57-58]。
有機質的存在會抑制Fe(Ⅱ) 催化鐵氧化物的相變反應。Jones 等人[55]證明富里酸抑制 Fe(Ⅱ) 催化鐵氧化物轉化為更穩定的礦物相,反應條件為1 mmol/L Fe(Ⅱ),C/Fe摩爾比約0.6和4.2;類似地,Pasakarnis[59]在1mmol/L Fe(Ⅱ),C/Fe 摩爾比為1和2條件下,發現OM可以防止Fe(Ⅱ) 催化 OM–水鐵礦共沉淀物轉化為磁鐵礦或針鐵礦。Chen等人[60]在研究Fe(Ⅱ) 催化有機物–水鐵礦共沉淀試驗中,發現有機物含量越高對水鐵礦相變的抑制作用越明顯,當C/Fe 摩爾比大于1.6時,水鐵礦表面的吸附位點幾乎被有機質占據,此時水鐵礦的晶體轉化完全被抑制,同時該研究還表明有機物的存在促使水鐵礦相變形成纖鐵礦。此外,有機質中存在多種官能團,Fe(Ⅱ)可以與這些官能團結合,從而降低了在鐵氧化物表面的吸附;另一方面,有機質吸附于鐵氧化物表面,能夠阻礙鐵氧化物的成核作用,從而抑制水鐵礦的晶相轉變[61]。
金屬陽離子的存在也會對 Fe(Ⅱ) 催化鐵氧化物相變有著重要影響。在此類研究中,二價金屬陽離子備受關注。例如,在一些二價陽離子如 Zn(Ⅱ)、Cu(Ⅱ) 和Mn(Ⅱ) 的存在下,由于離子半徑接近 Fe(Ⅱ),導致 Fe(Ⅱ) 催化水鐵礦形成了陽離子替代Fe(Ⅱ) 的磁鐵礦[62]。Liu等人[63]研究了Mg(Ⅱ)、Ca(Ⅱ)、Ba(Ⅱ)、Mn(Ⅱ)、Co(Ⅱ)、Ni(Ⅱ)和Zn(Ⅱ) 7種金屬陽離子對Fe(Ⅱ) 催化水鐵礦轉化過程的影響,結果發現陽離子與水鐵礦之間結合能力顯著影響水鐵礦的相變過程,具有較高結合能力的陽離子降低了水鐵礦上Fe(Ⅱ) 的吸附量,并降低了反應體系的氧化還原電位,從而抑制了水鐵礦的轉化;此外,這些離子也影響了水鐵礦相變的產物類型,在具有較低結合能力的陽離子存在時,水鐵礦先轉化為纖鐵礦,然后轉化為針鐵礦和磁鐵礦,而在具有更高結合能力的陽離子存在下,水鐵礦直接轉化為針鐵礦和磁鐵礦。除上述二價金屬陽離子,Al3+的作用也不容忽視,鐵氧化物發生鋁同晶替代是土壤中普遍的礦物學現象。Hansel等人[64]研究了Al替代以及Al吸附對Fe(Ⅱ) 催化水鐵礦相變的影響,發現Al替代以及Al吸附都會抑制水鐵礦的相變,Al替代或Al吸附量越高,抑制作用越強;同時,Fe(Ⅱ)催化水鐵礦相變產生的次生礦物類型也受控于Al替代或Al吸附量,發現Al取代會阻礙纖鐵礦形成和磁鐵礦成核,而Al 吸附完全抑制針鐵礦的形成,對磁鐵礦成核的影響較小。
4.3 亞鐵催化鐵氧化物相變的環境效應
鐵氧化物的晶相轉化,會導致其表面性質以及反應活性的改變,從而影響與其相關聯的重金屬和有機質在環境中的地球化學行為。亞鐵催化鐵氧化物的相變過程中,會導致與鐵氧化物共存的重金屬離子發生吸附解吸、氧化還原、共沉淀、摻雜等眾多反應,進而影響這些重金屬離子的遷移轉化。Pederen等人[65]研究發現,Fe(Ⅱ) 催化水鐵礦、纖鐵礦形成的新生鐵氧化物對砷酸鹽的吸附能力更強,認為缺氧條件下Fe(Ⅱ) 催化鐵氧化物重結晶可能是As固定的重要機制。Hu等人[66]發現Fe(Ⅱ) 催化水鐵礦相變過程中,溶液中的As摻雜進入新形成的礦物晶格內部。劉承帥等人[15]研究了Pb(Ⅱ) 共存下,Fe(Ⅱ) 催化水鐵礦相變過程,發現在水鐵礦晶相轉變過程中,部分吸附在水鐵礦表面的 Pb(Ⅱ) 通過晶體包裹或Fe結構位取代的方式被固定,從而降低了Pb(Ⅱ) 的移動性。Boland等人[67]研究表明Fe(Ⅱ) 催化水鐵礦相變過程中,會導致U(Ⅵ) 還原為U(Ⅴ) 并固定于新形成的針鐵礦結構中。而另一方面,上述這些被固定的重金屬會在鐵氧化物發生微生物或化學溶解時釋放到土壤環境中,造成相應的重金屬環境污染。
鐵氧化物很少單獨存在,常與環境中大量存在的有機質組成復合物。據估計,沉積物中大約21.5% 的有機碳與Fe通過共沉淀或螯合的形式共存[68],鐵氧化物與有機質組成的復合物能夠有效抑制微生物對有機物的分解,從而有利于環境中有機碳的固存。Fe(Ⅱ) 催化水鐵礦相變反應會間接影響有機質的保存,因為水鐵礦具有極大的比表面積,相比相變產生的高結晶度的鐵氧化物,水鐵礦有利于吸附或共沉淀更多的有機質。對此,Chen等[60]研究認為還原環境下,Fe(Ⅱ) 催化的水鐵礦相變過程可能會降低自然環境中的有機質的穩定性。此外,Fe(Ⅱ)催化水鐵礦相變也會間接影響微生物對鐵氧化物的利用性,通常,結晶度較低的水鐵礦更易于被微生物所還原利用。因此,Fe(Ⅱ) 催化水鐵礦相變形成結晶態鐵氧化物將會抑制微生物對鐵的還原利用,進而影響參與微生物鐵呼吸的元素的地球化學行為。
5 展望
目前對于鐵氧化物–亞鐵之間的相互作用的反應機制及其環境影響的室內研究取得了眾多好的結果與認識,但由于自然土壤環境條件復雜多變,參與反應的物質種類眾多,當前的研究尚有不足,在未來的研究中,以下幾個方面值得關注。
1)實際土壤沉積物環境中難以存在純的鐵氧化物,常會伴隨離子或分子的摻雜,且鐵氧化物常與有機質復合,而目前的研究僅限于實驗室合成的純鐵氧化物體系,需進一步模擬研究自然環境中鐵氧化物對Fe(Ⅱ) 氧化成礦的影響。另一方面,鐵氧化物存在下,Fe(Ⅱ) 氧化形成的新生鐵氧化物的性質,包括礦物結構、形貌、粒徑等礦物學特征以及生物化學反應活性,還需更加深入的研究。
2)目前關于Fe(Ⅱ) –鐵氧化物還原污染物以及Fe(Ⅱ) 催化鐵氧化物相變耦合重金屬遷移轉化的認識尚限于較為單一的實驗條件,如何將實驗所取得的認識應用于復雜動態的自然系統中,仍需擴展實驗反應參數,并基于熱力學和動力學數據,建立反應模型,模擬實際環境反應發生,預測污染物在土壤中的行為。
3)一些土壤環境存在重金屬與有機污染物組成的復合污染,目前關于研究Fe(Ⅱ)–鐵氧化物系統在復合污染物系統中的表現尚顯不足,需要進一步研究關注,以探索Fe(Ⅱ)–鐵氧化物系統在處理該類污染的可行性。
4)當前的研究大多以室內實驗室模擬反應為主,未來的研究需關注自然土壤中,鐵氧化物與溶液相亞鐵的界面反應及環境影響。鑒于實際土壤系統的復雜性,目前常規分析表征技術在應用于實地土壤界面反應微觀尺度的研究仍有眾多困難,需要開發更加適用于實際土壤環境的原位觀測技術手段和方法。
[1] Kappler A, Schink B, Newman D K. Fe(Ⅲ) mineral formation and cell encrustation by the nitrate-dependent Fe(Ⅱ)-oxidizer strain BoFeN1[J]. Geobiology, 2005, 3(4): 235–245.
[2] 胡敏, 李芳柏. 土壤微生物鐵循環及其環境意義[J]. 土壤學報, 2014, 51(4): 683–698.
[3] Buerge I J, Hug S J. Influence of mineral surfaces on chromium(Ⅵ) reduction by iron(Ⅱ)[J]. Environmental Science & Technology, 1999, 33(23): 4285–4291.
[4] Charlet L, Bosbach D, Peretyashko T. Natural attenuation of TCE, As, Hg linked to the heterogeneous oxidation of Fe(Ⅱ): An AFM study[J]. Chemical Geology, 2002, 190(1/2/3/4): 303–319.
[5] Amonette J E, Workman D J, Kennedy D W, et al. Dechlorination of carbon tetrachloride by Fe(Ⅱ) associated with goethite[J]. Environmental Science & Technology, 2000, 34(21): 4606–4613.
[6] Chun C L, Penn R L, Arnold W A. Kinetic and microscopic studies of reductive transformations of organic contaminants on goethite[J]. Environmental Science & Technology, 2006, 40(10): 3299–3304.
[7] Elsner M, Schwarzenbach R P, Haderlein S B. Reactivity of Fe(Ⅱ)-bearing minerals toward reductive transformation of organic contaminants[J]. Environmental Science & Technology, 2004, 38(3): 799–807.
[8] Jones A M, Kinsela A S, Collins R N, et al. The reduction of 4-chloronitrobenzene by Fe(Ⅱ)-Fe(Ⅲ) oxide systems - correlations with reduction potential and inhibition by silicate[J]. Journal of Hazardous Materials, 2016, 320: 143–149.
[9] Pecher K, Haderlein S B, Schwarzenbach R P. Reduction of polyhalogenated methanes by surface-bound Fe(Ⅱ) in aqueous suspensions of iron oxides[J]. Environmental Science & Technology, 2002, 36(8): 1734–1741.
[10] Tai Y L, Dempsey B A. Nitrite reduction with hydrous ferric oxide and Fe(Ⅱ): Stoichiometry, rate, and mechanism[J]. Water Research, 2009, 43(2): 546–552.
[11] Larese-Casanova P, Kappler A, Haderlein S B. Heterogeneous oxidation of Fe(Ⅱ) on iron oxides in aqueous systems: Identification and controls of Fe(Ⅲ) product formation[J]. Geochimica et Cosmochimica Acta, 2012, 91: 171–186.
[12] Williams A G B, Scherer M M. Spectroscopic evidence for Fe(Ⅱ)–Fe(Ⅲ) electron transfer at the iron oxide–water interface[J]. Environmental Science & Technology, 2004, 38(18): 4782–4790.
[13] Handler R M, Beard B L, Johnson C M, et al. Atom exchange between aqueous Fe(Ⅱ) and goethite: An Fe isotope tracer study[J]. Environmental Science & Technology, 2009, 43(4): 1102–1107.
[14] Yanina S V, Rosso K M. Linked reactivity at mineral-water interfaces through bulk crystal conduction[J]. Science, 2008, 320(5873): 218–222.
[15] 劉承帥, 李芳柏, 陳曼佳, 等. Fe(Ⅱ)催化水鐵礦晶相轉變過程中Pb的吸附與固定[J]. 化學學報, 2017, 75(6): 621–628.
[16] Tartaj P, Morales M P, Gonzalez-Carre?o T, et al. The iron oxides strike back: From biomedical applications to energy storage devices and photoelectrochemical water splitting[J]. Advanced Materials, 2011, 23(44): 5243–5249.
[17] Cornell R M, Schwertmann U. The iron oxides: Structure, properties, reactions, occurrences, and uses[M]. Weinheim: Wiley-VCH, 2003.
[18] Hiemstra T. Formation, stability, and solubility of metal oxide nanoparticles: Surface entropy, enthalpy, and free energy of ferrihydrite[J]. Geochimica et Cosmochimica Acta, 2015, 158: 179–198.
[19] Liao S, Wang X M, Yin H, et al. Effects of Al substitution on local structure and morphology of lepidocrocite and its phosphate adsorption kinetics[J]. Geochimica et Cosmochimica Acta, 2020, 276: 109–121.
[20] Chorover J, Amistadi M K. Reaction of forest floor organic matter at goethite, birnessite and smectite surfaces[J]. Geochimica et Cosmochimica Acta, 2001, 65(1): 95–109.
[21] Huang X P, Hou X J, Zhang X, et al. Facet-dependent contaminant removal properties of hematite nanocrystals and their environmental implications[J]. Environmental Science: Nano, 2018, 5(8): 1790–1806.
[22] Navrotsky A, Mazeina L, Majzlan J. Size-driven structural and thermodynamic complexity in iron oxides[J]. Science, 2008, 319(5870): 1635–1638.
[23] 胡世文. 鐵礦物生物和非生物轉化過程中砷和有機碳的微觀固存機制[D]. 廣州: 華南理工大學, 2021.
[24] Huang J Z, Jones A, Waite T D, et al. Fe(Ⅱ) redox chemistry in the environment[J]. Chemical Reviews, 2021, 121(13): 8161–8233.
[25] Melton E D, Swanner E D, Behrens S, et al. The interplay of microbially mediated and abiotic reactions in the biogeochemical Fe cycle[J]. Nature Reviews Microbiology, 2014, 12(12): 797–808.
[26] Morgan B, Lahav O. The effect of pH on the kinetics of spontaneous Fe(Ⅱ) oxidation by O2in aqueous solution - basic principles and a simple heuristic description[J]. Chemosphere, 2007, 68(11): 2080–2084.
[27] Park U, Dempsey B A. Heterogeneous oxidation of Fe(Ⅱ) on ferric oxide at neutral pH and a low partial pressure of O2[J]. Environmental Science & Technology, 2005, 39(17): 6494–6500.
[28] Tamura H, Kawamura S, Hagayama M. Acceleration of the oxidation of Fe2+ions by Fe(Ⅲ)-oxyhydroxides[J]. Corrosion Science, 1980, 20(8/9): 963–971.
[29] Jones A M, Griffin P J, Collins R N, et al. Ferrous iron oxidation under acidic conditions-The effect of ferric oxide surfaces[J]. Geochimica et Cosmochimica Acta, 2014, 145: 1–12.
[30] Chen C M, Thompson A. The influence of native soil organic matter and minerals on ferrous iron oxidation[J]. Geochimica et Cosmochimica Acta, 2021, 292: 254–270.
[31] Chen C M, Thompson A. Ferrous iron oxidation under varying pO2levels: The effect of Fe(Ⅲ)/Al(Ⅲ) oxide minerals and organic matter[J]. Environmental Science & Technology, 2018, 52(2): 597–606.
[32] Zwank L, Elsner M, Aeberhard A, et al. Carbon isotope fractionation in the reductive dehalogenation of carbon tetrachloride at iron (hydr)oxide and iron sulfide minerals[J]. Environmental Science & Technology, 2005, 39(15): 5634–5641.
[33] Maithreepala R A, Doong R A. Synergistic effect of copper ion on the reductive dechlorination of carbon tetrachloride by surface-bound Fe(Ⅱ) associated with goethite[J]. Environmental Science & Technology, 2004, 38(1): 260–268.
[34] Colón D, Weber E J, Anderson J L. Effect of natural organic matter on the reduction of nitroaromatics by Fe(Ⅱ) species[J]. Environmental Science & Technology, 2008, 42(17): 6538–6543.
[35] Luan F B, Xie L, Li J, et al. Abiotic reduction of nitroaromatic compounds by Fe(Ⅱ) associated with iron oxides and humic acid[J]. Chemosphere, 2013, 91(7): 1035–1041.
[36] Jeon B H, Dempsey B A, Burgos W D, et al. Chemical reduction of U(Ⅵ) by Fe(Ⅱ) at the solid-water interface using natural and synthetic Fe(Ⅲ) oxides[J]. Environmental Science & Technology, 2005, 39(15): 5642–5649.
[37] Peretyazhko T, Zachara J M, Heald S M, et al. Heterogeneous reduction of Tc(Ⅶ) by Fe(Ⅱ) at the solid-water interface[J]. Geochimica et Cosmochimica Acta, 2008, 72(6): 1521–1539.
[38] Jang J H, Dempsey B A, Burgos W D. Reduction of U(Ⅵ) by Fe(Ⅱ) in the presence of hydrous ferric oxide and hematite: Effects of solid transformation, surface coverage, and humic acid[J]. Water Research, 2008, 42(8/9): 2269–2277.
[39] Hering J, Stumm W. Oxidative and reductive dissolution of minerals[J]. Reviews in Mineralogy & Geochemistry, 1990, 23: 427–465.
[40] Orsetti S, Laskov C, Haderlein S B. Electron transfer between iron minerals and quinones: Estimating the reduction potential of the Fe(Ⅱ)-goethite surface from AQDS speciation[J]. Environmental Science & Technology, 2013, 47(24): 14161–14168.
[41] Silvester E, Charlet L, Tournassat C, et al. Redox potential measurements and M?ssbauer spectrometry of FeⅡadsorbed onto FeⅢ(oxyhydr)oxides[J]. Geochimica et Cosmochimica Acta, 2005, 69(20): 4801–4815.
[42] Gorski C A, Scherer M M. Influence of magnetite stoichiometry on Fe(Ⅱ) uptake and nitrobenzene reduction[J]. Environmental Science & Technology, 2009, 43(10): 3675–3680.
[43] Larese-Casanova P, Scherer M M. Fe(Ⅱ) sorption on hematite: New insights based on spectroscopic measurements[J]. Environmental Science & Technology, 2007, 41(2): 471–477.
[44] Rosso K M, Yanina S V, Gorski C A, et al. Connecting observations of hematite (α-Fe2O3) growth catalyzed by Fe(Ⅱ)[J]. Environmental Science & Technology, 2010, 44(1): 61–67.
[45] Felmy A R, Ilton E S, Rosso K M, et al. Interfacial reactivity of radionuclides: Emerging paradigms from molecular-level observations[J]. Mineralogical Magazine, 2011, 75(4): 2379–2391.
[46] Felmy A R, Moore D A, Rosso K M, et al. Heterogeneous reduction of PuO2with Fe(Ⅱ): Importance of the Fe(Ⅲ) reaction product[J]. Environmental Science & Technology, 2011, 45(9): 3952–3958.
[47] Stemig A M, Do T A, Yuwono V M, et al. Goethite nanoparticle aggregation: Effects of buffers, metal ions, and 4-chloronitrobenzene reduction[J]. Environmental Science: Nano, 2014, 1(5): 478–487.
[48] Cwiertny D M, Handler R M, Schaefer M V, et al. Interpreting nanoscale size-effects in aggregated Fe-oxide suspensions: Reaction of Fe(Ⅱ) with goethite[J]. Geochimica et Cosmochimica Acta, 2008, 72(5): 1365–1380.
[49] Zhang Y, Charlet L, Schindler P W. Adsorption of protons, Fe(Ⅱ) and Al(Ⅲ) on lepidocrocite (γ-FeOOH)[J]. Colloids and Surfaces, 1992, 63(3/4): 259–268.
[50] Liger E, Charlet L, Van Cappellen P. Surface catalysis of uranium(Ⅵ) reduction by iron(Ⅱ)[J]. Geochimica et Cosmochimica Acta, 1999, 63(19/20): 2939–2955.
[51] Charlet L, Silvester E, Liger E. N-compound reduction and actinide immobilisation in surficial fluids by Fe(Ⅱ): The surface FeⅢOFeⅡOH° species, as major reductant[J]. Chemical Geology, 1998, 151(1/2/3/4): 85–93.
[52] Zhang H C, Taujale S, Huang J Z, et al. Effects of NOM on oxidative reactivity of manganese dioxide in binary oxide mixtures with goethite or hematite[J]. Langmuir: the ACS Journal of Surfaces and Colloids, 2015, 31(9): 2790–2799.
[53] Vindedahl A M, Stemig M S, Arnold W A, et al. Character of humic substances as a predictor for goethite nanoparticle reactivity and aggregation[J]. Environmental Science & Technology, 2016, 50(3): 1200–1208.
[54] 胡世文, 劉同旭, 李芳柏, 等. 土壤鐵礦物的生物-非生物轉化過程及其界面重金屬反應機制的研究進展[J]. 土壤學報, 2022, 59(1): 54–65.
[55] Jones A M, Collins R N, Rose J, et al. The effect of silica and natural organic matter on the Fe(Ⅱ)-catalysed transformation and reactivity of Fe(Ⅲ) minerals[J]. Geochimica et Cosmochimica Acta, 2009, 73(15): 4409–4422.
[56] Boland D D, Collins R N, Miller C J, et al. Effect of solution and solid-phase conditions on the Fe(Ⅱ)-accelerated transformation of ferrihydrite to lepidocrocite and goethite[J]. Environmental Science & Technology, 2014, 48(10): 5477–5485.
[57] Hansel C M, Benner S G, Fendorf S. Competing Fe (Ⅱ)-induced mineralization pathways of ferrihydrite[J]. Environmental Science & Technology, 2005, 39(18): 7147–7153.
[58] Hansel C M, Benner S G, Neiss J, et al. Secondary mineralization pathways induced by dissimilatory iron reduction of ferrihydrite under advective flow[J]. Geochimica et Cosmochimica Acta, 2003, 67(16): 2977–2992.
[59] Pasakarnis T S. Effects of carbon during Fe(Ⅱ)-catalyzed Fe oxide recrystallization[D]. The University of Iowa, Doctor of Philosophy, 2013. DOI: 10.17077/etd.wnhgjk73
[60] Chen C M, Kukkadapu R, Sparks D L. Influence of coprecipitated organic matter on Fe2+(aq)-catalyzed transformation of ferrihydrite: Implications for carbon dynamics[J]. Environmental Science & Technology, 2015, 49(18): 10927–10936.
[61] Xiao W, Jones A M, Collins R N, et al. Investigating the effect of ascorbate on the Fe(Ⅱ)-catalyzed transformation of the poorly crystalline iron mineral ferrihydrite[J]. Biochimica et Biophysica Acta (BBA) - General Subjects, 2018, 1862(8): 1760–1769.
[62] Jang J H, Dempsey B A, Catchen G L, et al. Effects of Zn(Ⅱ), Cu(Ⅱ), Mn(Ⅱ), Fe(Ⅱ), NO3?, or SO42?at pH 6.5 and 8.5 on transformations of hydrous ferric oxide (HFO) as evidenced by M?ssbauer spectroscopy[J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2003, 221(1/2/3): 55–68.
[63] Liu C S, Zhu Z K, Li F B, et al. Fe(Ⅱ)-induced phase transformation of ferrihydrite: The inhibition effects and stabilization of divalent metal cations[J]. Chemical Geology, 2016, 444: 110–119.
[64] Hansel C M, Learman D R, Lentini C J, et al. Effect of adsorbed and substituted Al on Fe(Ⅱ)-induced mineralization pathways of ferrihydrite[J]. Geochimica et Cosmochimica Acta, 2011, 75(16): 4653–4666.
[65] Pedersen H D, Postma D, Jakobsen R. Release of arsenic associated with the reduction and transformation of iron oxides[J]. Geochimica et Cosmochimica Acta, 2006, 70(16): 4116–4129.
[66] Hu S W, Lu Y, Peng L F, et al. Coupled kinetics of ferrihydrite transformation and As(Ⅴ) sequestration under the effect of humic acids: A mechanistic and quantitative study[J]. Environmental Science & Technology, 2018, 52(20): 11632–11641.
[67] Boland D D, Collins R N, Glover C J, et al. Reduction of U(Ⅵ) by Fe(Ⅱ) during the Fe(Ⅱ)-accelerated transformation of ferrihydrite[J]. Environmental Science & Technology, 2014, 48(16): 9086–9093.
[68] Lalonde K, Mucci A, Ouellet A, et al. Preservation of organic matter in sediments promoted by iron[J]. Nature, 2012, 483(7388): 198–200.
Soil Iron Oxide-Ferrous Interaction and Its Environmental Effects: A Review
YAO Yuan1,2, YU Guanghui1, TENG Hui1*
(1Institute of Surface-Earth System Science, School of Earth System Science, Tianjin University, Tianjin 300072, China; 2 Xi’an Center of China Geological Survey, Xi’an 710119, China)
Iron (hydr)oxides (hereafter termed iron oxides) catalyzed oxidation of aqueous Fe(Ⅱ) is an important process in soil environments not only because the two phases often coexist but also because the reactions are frequently coupled with the reduction of organic and inorganic contaminants. Concurrently, recrystallization of iron oxides in the presence of aqueous Fe(Ⅱ) plays a critical part in the iron geochemical cycling as the process leads to changes in the structure and surface properties of iron oxides to alter the ensuing adsorption of heavy metals and organic matter in soils and sediments. In this paper, we reviewed the recent progresses in understanding the interactions of iron oxides and aqueous ferrous with a focus on the reaction mechanism and controlling factors for: 1) iron oxide-catalyzed Fe(Ⅱ) oxidation; 2) contaminants reduction by the aqueous Fe(Ⅱ)-iron oxides system; and 3) Fe(Ⅱ)-catalyzed recrystallization of iron oxides. In addition, we provide a perspective for the future research of the iron oxide-aqueous Fe(Ⅱ) interface reaction and related environmental impact in soils.
Iron oxide; Fe(Ⅱ) oxidation; Mineral transformation; Organic contaminants; Heavy metals
X142
A
10.13758/j.cnki.tr.2023.04.004
姚遠, 余光輝, 滕輝. 土壤鐵氧化物–亞鐵的相互作用及其環境影響研究進展. 土壤, 2023, 55(4): 718–728.
國家自然科學基金重點項目(41830859)資助。
(huihenry.teng@tju.edu.cn)
姚遠(1997—),男,陜西西安人,碩士,主要研究方向為土壤鐵礦物的形成及其環境效應。E-mail: YaoYuanSF@163.com
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
