基團辨識加氫:從概念到應用
2023-10-07 12:34:26毛善俊王哲王勇
化工進展 2023年8期
關鍵詞:催化劑
毛善俊,王哲,王勇
(浙江大學化學系催化研究所,能源高效清潔利用全國重點實驗室,化學前瞻技術研究中心,浙江 杭州 310058)
催化加氫在生產大宗化學品和精細化學品領域中發揮著舉足輕重的作用。據統計,約有25%的化學過程涉及加氫反應。選擇性加氫是最具挑戰性的反應類型之一。當反應物中存在多種可還原官能團時(或者存在串聯加氫反應),需要優先還原特定官能團,以生產高價值的化學品。然而,某些催化劑在提高選擇性的同時犧牲了活性,這種設計策略已經難以滿足化工產業轉型升級的需求。顯然,催化劑對不同基團的響應程度將決定多種可還原官能團的轉化效率,并最終表現出不同的反應選擇性。盡管許多研究都致力于調節加氫反應的選擇性,但由于對活性位點與官能團之間的相互作用缺乏更深入的了解,實現基團的定向轉化仍然具有極大挑戰性。為了合理設計更高效的多相催化選擇性加氫催化劑,實現反應過程的清潔化、綠色化,本文在前期工作的基礎上提煉出“基團辨識加氫”的概念,旨在為該領域的快速健康發展提供新的解決思路。
1 “基團辨識加氫”概念的內涵
負載型納米金屬催化劑是最重要的加氫催化劑之一,在工業上應用十分廣泛。過渡金屬納米顆粒具有較寬的d 帶,通常同時具備活潑電子和空的軌道能級。因此,當它們充當反應活性位點時,既可作為電子給體,也可以作為電子受體。這使得催化劑表現出多種反應活性,從而導致加氫選擇性受到較大程度的制約。傳統選擇性加氫主要指多個可還原官能團共存時,對特定官能團的優先還原轉化,強調同一時空下的優先性。催化劑的設計有時以犧牲活性作為代價換取較高的選擇性。而“基團辨識加氫”則以催化劑對目標基團的定向活化或辨識響應為原則,要求在提高反應選擇性的同時不犧牲活性,更注重活性位對目標基團的“一對一”特異性響應,從而突破了時空的限制。這一概念對傳統選擇性加氫進行了深化,并對催化劑設計提出了更高的要求,同時期望實現更優的催化性能[1]。一個典型的例子是,如果某一活性位對基團A 和B 都具有加氫活性,但是在A 和B 同時存在條件下能夠優先轉化其中一種基團,這種情況可以被稱為選擇性加氫。然而,在基團辨識加氫的理想情況下,活性位只能對其中一種基團具有響應或加氫活性。
2 實現“基團辨識加氫”的策略
2.1 調節活性位點和目標基團之間的軌道對稱性和能級匹配度,從能量空間實現調控
催化劑活性位點和目標基團之間的相互作用需要滿足成鍵三原則,即能量近似、對稱性匹配和軌道最大重疊原則,才能實現有效活化。其中,能量近似和最大重疊原則決定成鍵的效率,而對稱性匹配決定了是否能夠成鍵。對于給定的目標基團,其與活性位點的軌道對稱性和軌道能級可以認為是獨立變量,而最大重疊原則、軌道對稱性和能級之間存在密切關系。因此,活性位點和目標基團之間的軌道對稱性和能級匹配度是調節兩者相互作用的關鍵因素。以鹵代硝基苯選擇性加氫制鹵代苯胺的反應為例,該反應對負載型納米金屬催化劑具有結構敏感性,低配位邊角位點容易引起脫鹵副反應。納米金屬顆粒尺寸越小,脫鹵副反應越嚴重。這意味著納米金屬的顆粒尺寸需要足夠大才能維持較高的選擇性,但這也導致貴金屬利用率較低。同時,在反應過程中,通常還需要添加有機助劑來進一步毒化催化劑,抑制副反應,通過犧牲活性以達到工業要求的選擇性。
通過對氯代苯胺分子的前線軌道進行分析,本文作者課題組發現C—Cl的非鍵軌道和貴金屬活性位點的d軌道之間的對稱性匹配程度是決定是否發生脫鹵副反應的關鍵[2]。如圖1 所示,由于C—Cl非鍵軌道的雙象限分布特點,而低配位邊角位點由于暴露的金屬d 軌道也具有雙象限特征(dxz和dyz),因此可以與C—Cl 的非鍵軌道進行匹配,導致脫鹵副反應的發生。相反,高配位平面位點暴露的d軌道具有很強的單象限dz2軌道特性,對C—Cl的活化沒有響應。基于以上認識,本文作者課題組設計了單位點Pt1/CeO2催化劑,利用氧配位原子為金屬的雙象限金屬d軌道配置了兩個相反象限的軌道,改變了原有暴露d軌道的對稱性。該催化劑的Pt單位點對C—Cl顯示本征惰性,因而表現出大于99%的鹵代苯胺選擇性。同時,貴金屬理論原子利用率從小于10%提升至100%,表觀活性提升了26倍。類似的,通過調控金屬位點暴露的d軌道的對稱性,強化d 軌道電子的自旋極化,成功實現Co位點在C= = C 雙鍵存在條件下對硝基的辨識加氫。這種調控使得Co 位點對氨基苯乙烯顯示出選擇性大于99%[3]。
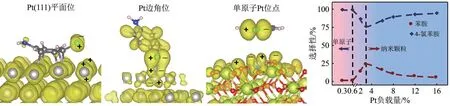
圖1 C—Cl基團非鍵軌道和Pt活性位點d軌道的對稱性匹配原理以及相應的加氫性能對比[2]
當軌道對稱性匹配條件滿足時,調控活性位點與目標基團的能級匹配度也可以調節兩者的相互作用,實現辨識響應。同樣以碳鹵鍵活化為例,目標基團的分子軌道能級是確定的,因此只能通過調節活性位點的能級來實現能級匹配度的調控[4]。理論計算結果表明[圖2(a)],催化劑的費米能級通常由納米金屬活性位提供,并受載體的功函影響。當催化劑載體從不可還原性載體Al2O3變為可還原性載體CeO2時,金屬位點的費米能級與C—Cl反鍵軌道的能級差從2.86eV 擴大至3.71eV。這個增大的能級差降低了金屬d 軌道向C—Cl 反鍵軌道(p*)注入電子的能力,從而顯著降低了脫鹵活性。實驗結果也證實了這一點[圖2(b)],相同形貌和尺寸的Pt納米顆粒脫鹵能力和載體功函呈線性關系。載體功函絕對值越大,抑制脫鹵能力越強。例如,2.6nm Pt 納米顆粒對4-氯苯胺的選擇性從Pt/γ-Al2O3的71%提升至Pt/CeO2的93%。
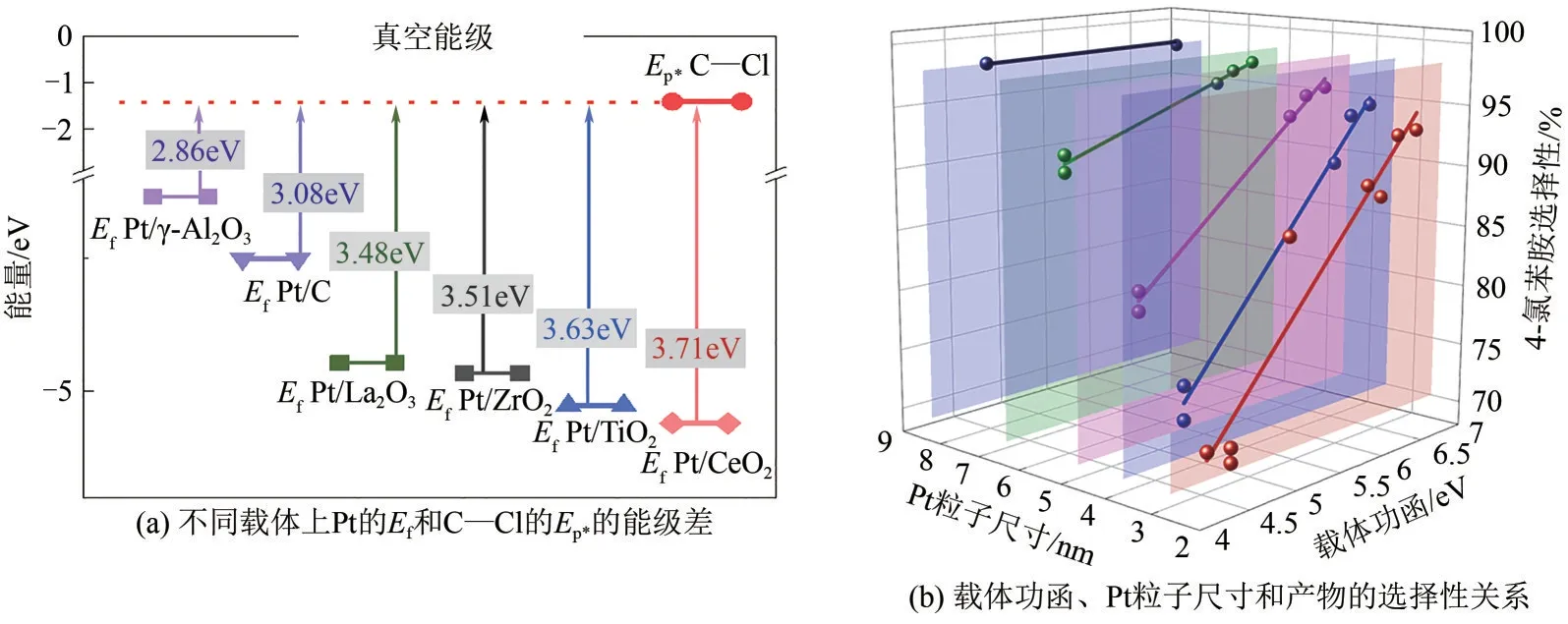
圖2 C—Cl基團反鍵軌道p*和Pt活性位點d軌道的能級匹配原理以及相應的加氫性能對比[4]
2.2 調節催化劑幾何結構,促進基團或底物分子與催化劑的空間結構適配,從物理空間實現調控
針對炔烴半加氫反應(圖3),Pd 表面不同的活性位對C≡C 和C= = C 均有反應活性,因此某些催化劑調控烯烴選擇性以犧牲活性為代價。理論計算研究表明,烯烴在Pd 邊角位點的強吸附是導致過加氫的重要原因,因此本文作者課題組采用靶向去活策略,利用金屬-載體強相互作用,控制催化劑的還原溫度,優先覆蓋Pd 顆粒表面能量高的邊角位,同時保留高選擇性的平面位點。通過這種策略,在Pd顆粒表面實現了對炔烴的辨識吸附,使得室溫下2-甲基-3-丁炔-2-醇的轉化頻率(TOFMBY)達到5526h-1,比商業Lindlar 催化劑提高了13 倍,且烯烴收率>97%[5-6]。此外,催化劑采用了孤島式結構,確保了高度的穩定性,實現了無鉛無有機助劑下液相炔烴半加氫的工業應用。
除了關注活性位和目標活性基團之間的相互作用以外,反應分子與催化劑載體的空間結構適配性也對催化劑的催化效率產生顯著影響。以長鏈共軛雙烯酮選擇性加氫制備飽和酮為例(圖4),該類分子的動力學直徑大(約0.71nm)、尺寸長(>2nm),傳統Pd/AC 催化劑由于微孔(<2nm)限制,導致內擴散慢,加氫反應表觀活性低,催化選擇性差。此外,由于兩個C= = C和一個C= = O形成的共軛結構不穩定,易產生低聚物并堵塞孔道,導致催化劑快速失活,循環使用性差,無法滿足工業生產的要求[7]。導致以上問題的直接原因是微孔傳質通道和大體積長鏈烷基基團的空間結構失配,影響了反應物以及活性中間體的傳質,進而導致過加氫和聚合副反應的加劇。因此,優化反應位點傳質通道的空間結構,構建具有大孔和介孔孔道的催化劑載體,將努森擴散轉變為表面擴散,實現催化劑對大分子的嚴苛擴散要求的辨識,是催化劑設計的關鍵。基于此,本文作者課題組開發“發泡法”制備具有高介孔和大孔比例的、開放多級孔道結構的氮摻雜多級孔炭(NHPC)。以NHPC為新型載體制備的負載型納米鈀催化劑(Pd/NHPC)可使目標產物收率高達99%,催化劑穩定套用130 次以上[7]。長鏈共軛雙烯酮辨識加氫關鍵技術的突破,開辟了全新的維生素E 側鏈異植物醇生產綠色工藝路線[1]。與國際同行技術相比,反應步驟由14 步減少到11 步,原子經濟性由36%提升至85%以上。
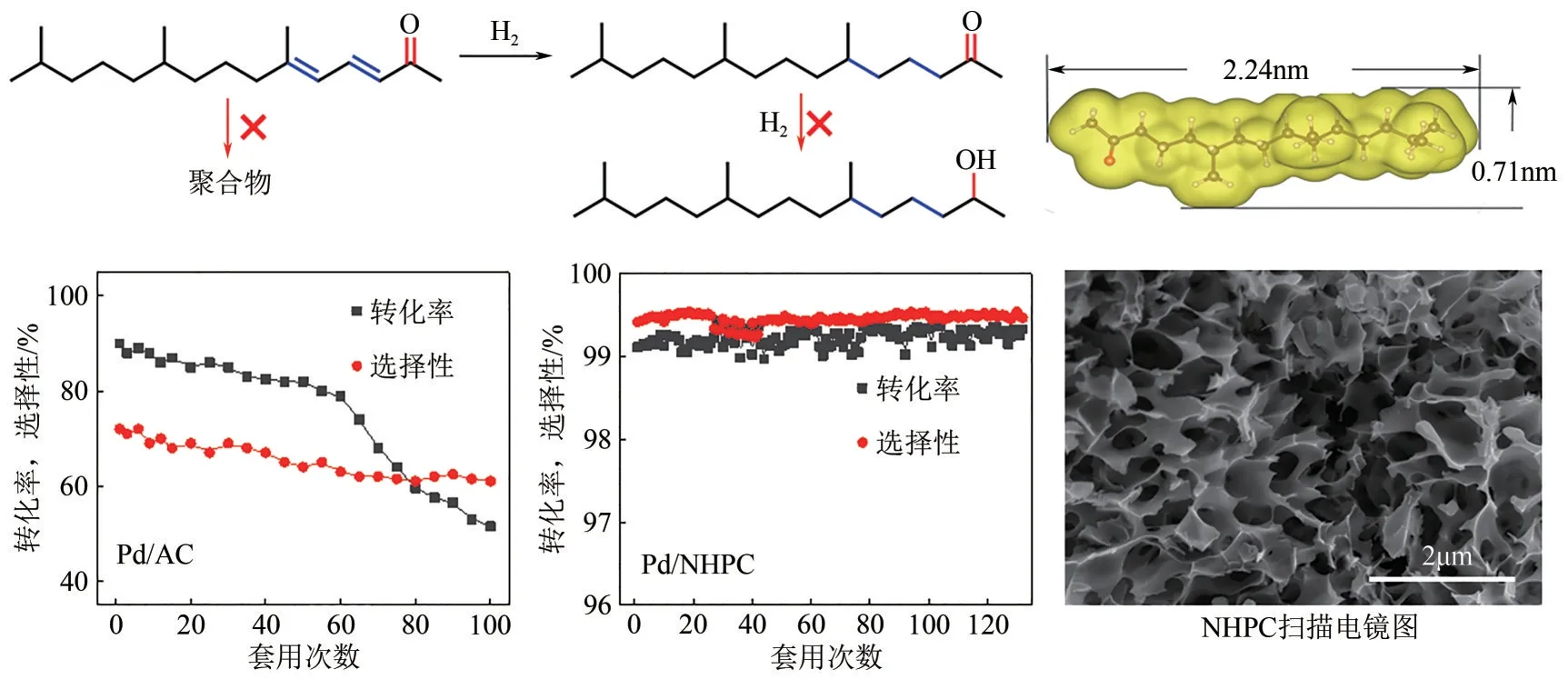
圖4 孔道和長鏈共軛雙烯酮空間結構適配程度和加氫性能對比[1]
2.3 催化劑活性位微環境調控實現基團辨識吸附和活化
活性位微環境調控也是實現辨識加氫的有效方法之一。以苯酚選擇性加氫為例,這是一類結構不敏感反應,Pd 表面不同類型的活性位對苯酚和目標產物環己酮均具有反應活性,導致環己酮容易發生串聯加氫生成環己醇。由于環己酮和環己醇對水的親和性不同,本文作者課題組利用氮摻雜增強炭材料的親水性,抑制了環己酮在Pd 表面的吸附,成功將環己酮選擇性提升至99%以上[8]。對炔烴半加氫反應,本文作者課題組首創“呼吸式”Pd 催化劑[圖5(a)]。該催化劑將長鏈柔性有機胺固載到炭載體表面,有機胺的柔性鏈采用可逆的“呼吸模式”改變了C≡C 和C= = C 的吸附熱力學特性,在吸入炔烴的同時選擇性地從Pd 表面排出烯烴,實現了對C≡C 的辨識吸附,從而抑制過加氫副反應,烯烴收率>99%,室溫下TOFMBY高達14412h-1,比商業Lindlar催化劑提高了35倍[9]。將上述呼吸式催化劑應用于以甲酸為氫源的生物質加氫脫氧反應[圖5(b)],Pd-有機胺微納功能基元高效分解甲酸生成高活性氫負離子(H-)對極性C= = O 鍵進行親核加成,實現了生物質混合含氧單體中芐基酮類單體分子的高效辨識轉化。常溫常壓條件下,反應活性比傳統高溫高壓的氫氣體系高一個數量級[10]。由于甲酸可以從生物質常溫水解得來,因此本文作者課題組提出了室溫條件下僅以生物質為原料合成燃料或高值化學品的高效“零碳”綠色合成路線。
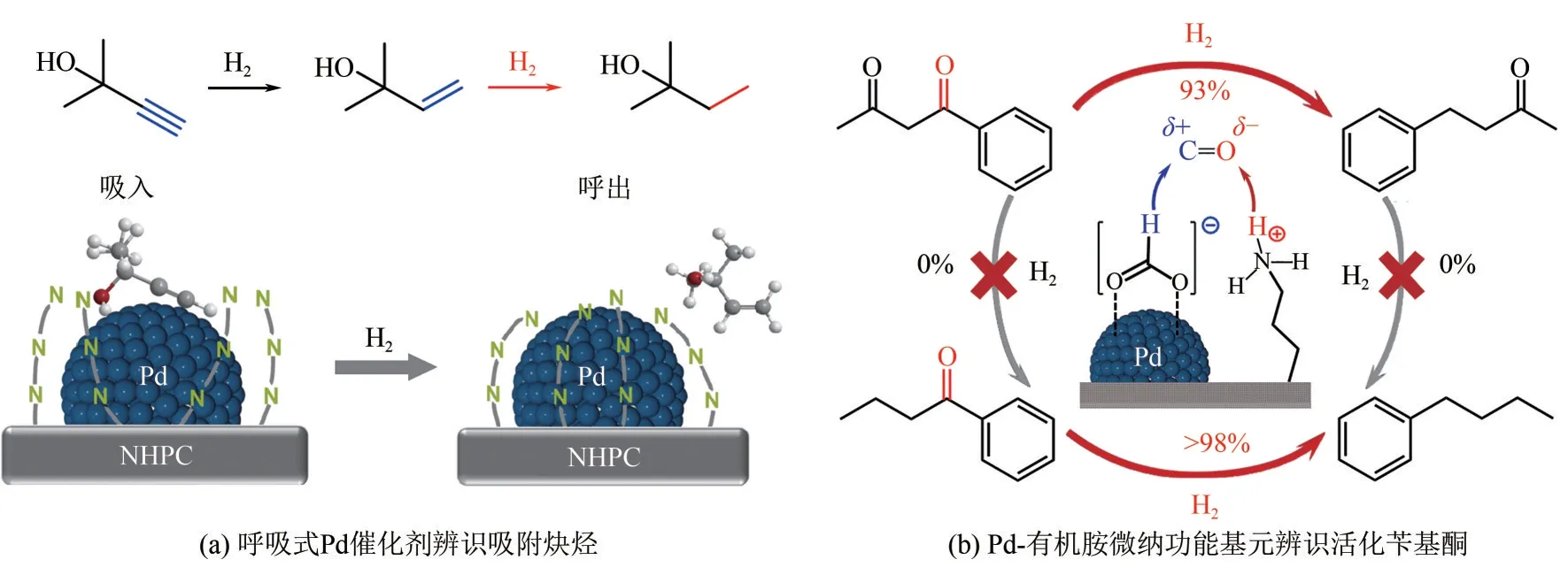
圖5 活性位微環境調控舉例實現辨識吸附和活化[9-10]
3 “基團辨識加氫”量化解析方法學基礎
“基團辨識加氫”概念的本質仍然是催化劑的電子和幾何結構調控,因此發展催化劑電子和幾何效應的量化評價手段至關重要。然而,電子和幾何效應往往互相耦合,難以拆分和定量解析。為盡量降低活性金屬幾何結構的影響,本文作者課題組采用膠體法和低溫活性氫置換法制備得到表面清潔的系列形貌相近、粒徑均一的負載型Pt 納米金屬催化劑[4]。實驗結果表明[圖6(a)],在對氯硝基苯選擇性加氫反應中,對氯苯胺的選擇性與不同載體上的零價Pt 含量呈線性關系,且該線性關系的斜率在不同Pt 顆粒粒徑下保持不變。由于Pt 顆粒幾何結構的影響已經被預先排除,這說明該線性關系代表了載體對該反應的電子效應,而由Pt 顆粒本身粒徑變化導致的電子結構變化在所研究的尺寸范圍內并不影響C—Cl鍵的活化。通過線性關系的斜率可進一步量化載體電子效應的強度。考慮到電子和幾何效應在數學上的正交關系,本文作者課題組首創正交分解法,以載體電子效應曲線為底作垂線,得到幾何效應曲線及其斜率,建立新二維直角坐標系,成功解耦了電子和幾何效應分別對加氫反應的相對貢獻。該方法還可推廣至三維直角坐標系[圖6(b)],在這種情況下,傳統尺寸效應和電子效應實際上是特殊視角的投影[1]。通過以上研究成果,本文作者課題組突破了“基團辨識加氫”構-效關系定量解析的瓶頸,為多相催化由經驗學科向解析學科轉變提供了一定的方法學基礎。
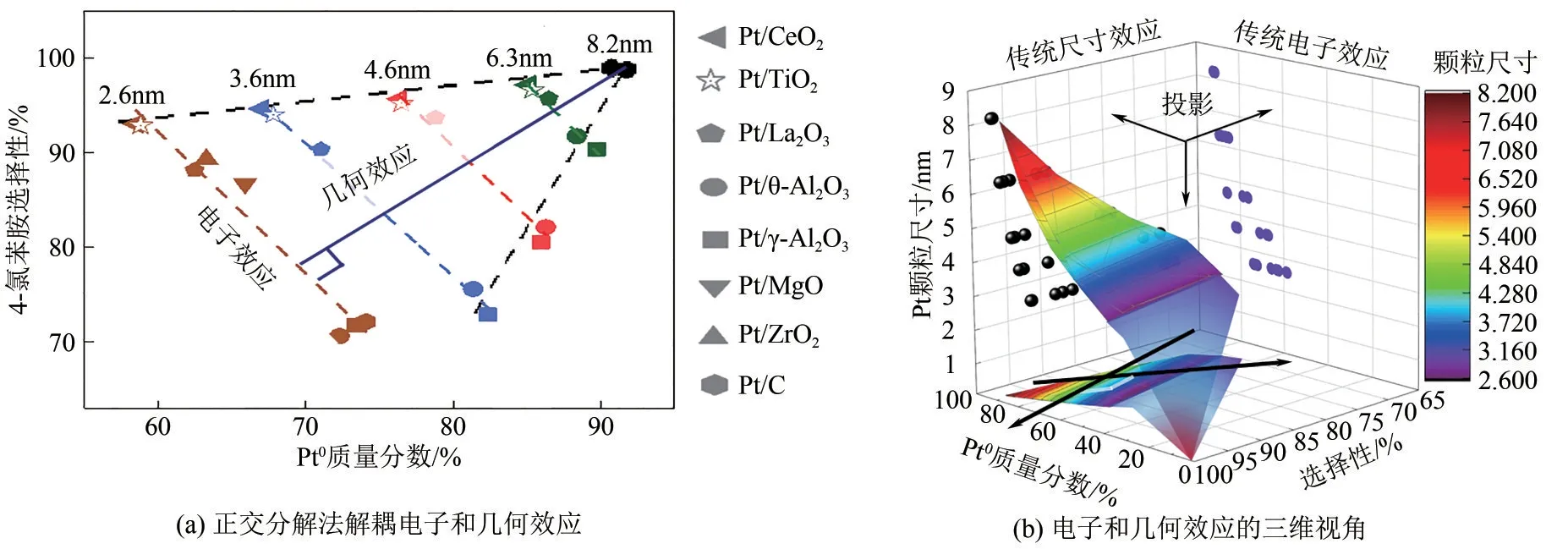
圖6 “基團辨識加氫”量化解析基本原理[4]
4 結語與展望
催化選擇性加氫在石油和精細化工等領域具有重要地位,是制備大宗和精細化學品及其中間體的綠色經濟途徑,同時也是提升我國化工產品精細化水平和國際競爭力的關鍵因素。而“基團辨識加氫”這一概念為選擇性加氫技術的發展帶來了新的思路。通過發展催化構-效關系定量解析方法學,“基團辨識加氫”有望顛覆現有的催化劑設計思想,引領多相催化領域由經驗學科向解析學科轉變,從而改變現有多相催化技術格局。這一新的思路和方法將為實現更高的催化選擇性和效率提供更可行的途徑,并為探索新型催化劑、優化反應條件和提高產物質量提供更深入的理論指導。因此,“基團辨識加氫”概念的提出對于催化科學和工程領域具有重要意義,有望推動催化領域的創新和發展。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
