鑭鈷鈣鈦礦催化劑制備及其催化氧化甲苯性能與機理
2023-10-12 02:44:40趙安蓮,任業為,曲振平
大連理工大學學報 2023年5期
關鍵詞:催化劑
趙 安 蓮, 任 業 為, 曲 振 平
(大連理工大學 環境學院 工業生態與環境工程教育部重點實驗室,遼寧 大連 116024 )
0 引 言
目前我國正在應對嚴重的臭氧污染和細顆粒物污染[1].臭氧和細顆粒物最重要的前驅體之一——揮發性有機化合物(volatile organic compounds,VOCs)[2],不僅會對生產生活造成影響,而且會對人體健康產生不同程度的損害[3].目前,加強對VOCs的處理是減少空氣污染的有效策略[4],而甲苯作為一種典型的揮發性有機化合物,其排放總量和臭氧形成潛力都位于前列[5],因此,消減和去除甲苯對于解決大氣污染有著重要意義.
催化氧化法通過使氣相有機廢氣與氧氣接觸,在催化劑作用下將VOCs轉化成完全無毒的小分子無機化合物[6],它具有反應溫度低、能耗低、適用范圍廣、二次污染物少等優點,被認為是去除VOCs最適宜有效的方法之一[7].該方法的關鍵在于催化劑的選擇與構建,不僅要考慮積炭或催化劑中毒等情況[8],還要考慮實際應用中的成本問題.因此,開發高效、穩定、低成本的催化劑對甲苯的催化氧化處理至關重要[7].
在眾多應用于消除VOCs的催化劑中,過渡金屬氧化物催化劑催化氧化VOCs的性能經過調控后能夠媲美貴金屬催化劑[9],且不易失活、中毒,成本更低廉,有著廣泛的應用前景[10].鈣鈦礦催化劑作為過渡金屬氧化物催化劑中的一種,其結構由ABO3表示,其中的A位元素一般為稀土、堿金屬或堿土金屬元素,而B位元素一般為過渡金屬元素[10].鈣鈦礦催化劑因其組成元素廣泛、制備方法簡單、價格低廉、結構可調變等優點,被認為是參與多相催化中貴金屬催化劑的替代品之一[11].然而,鈣鈦礦材料制備溫度[12]、催化氧化VOCs的活性及構效關系的相關探究仍有待完善[13].
本研究使用溶膠凝膠法制備LaCoO3催化劑,通過調控制備溫度優化LaCoO3催化劑的結構與性質,以期其在應用于催化氧化甲苯時具有較高的低溫轉化率,并結合原位表征手段探究相關機理.
1 實驗材料與方法
1.1 實驗材料
本研究使用的化學試劑與氣體見表1.

表1 化學試劑與氣體
1.2 鑭鈷鈣鈦礦催化劑的調控制備和表征
首先,將所需硝酸鹽前體La(NO3)3·6 H2O、Co(NO3)2·6 H2O和10 mmol的檸檬酸一水合物,根據1∶1∶2的物質的量比投加到500 mL燒杯中,并在磁力攪拌下溶解于50 mL去離子水中.然后,繼續將混合得到的溶膠前體物質在磁力攪拌下保持一定溫度直至形成凝膠,后轉移到100 ℃烘箱內進行發泡干燥保溫處理12 h,以得到疏松多孔的干凝膠.將干凝膠研磨成粉末并放入帶蓋小坩堝中,置于馬弗爐內以2 ℃/min的升溫速率從室溫加熱至煅燒溫度,并保持3 h,后自然冷卻至室溫以獲得黑灰色蓬松粉末,活性測試和質譜實驗前對粉末進行壓片過篩(20~40目)處理.
在本研究中,先探究溶膠凝膠形成溫度對得到的催化劑催化氧化甲苯活性的影響,在制備過程中,分別選擇在70、80、90和100 ℃下進行磁力攪拌直至形成凝膠,并統一選擇800 ℃作為馬弗爐內膽溫度進行煅燒,將得到的催化劑分別命名為SG-70、SG-80、SG-90和SG-100.
再探究煅燒溫度對得到的催化劑催化氧化甲苯活性的影響.保持在最佳溶膠凝膠形成溫度下進行磁力攪拌直至形成凝膠,分別將前體物質在500、600、700和800 ℃下進行煅燒,將最終得到的催化劑分別命名為CAL-500、CAL-600、CAL-700和CAL-800.
催化劑樣品物相由X射線衍射儀(XRD,Rigaku D/MAX-RB)測試分析得到.
1.3 催化氧化甲苯活性測試
催化氧化甲苯活性測試是在自行搭建的固定床微反應器上進行的,具體實驗裝置如圖1所示.
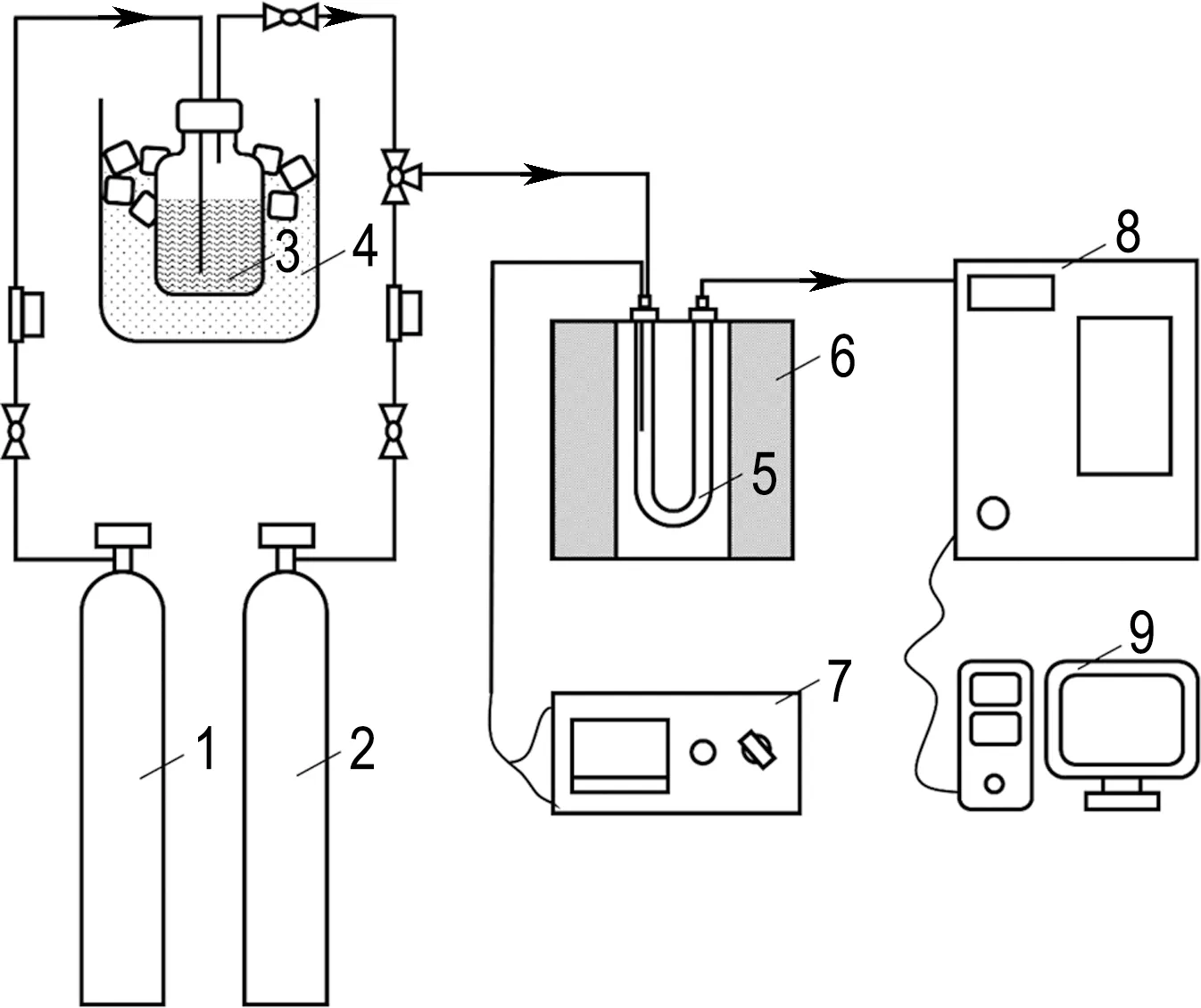
1 Ar; 2 O2/Ar; 3 液態甲苯; 4 冰水混合物; 5 石英反應管; 6 加熱爐; 7 溫度控制儀; 8 氣相色譜儀; 9 計算機
將Ar引入裝有液態甲苯的容器中,該容器浸沒在0 ℃冰水浴中,以保持生成的氣態甲苯濃度恒定.氣體混合物(436 mg/L甲苯+20% O2/Ar平衡)以75 mL/min的體積流量恒定通過石英反應管(內徑4 mm),空速為22 500 mL/(g·h),氣體混合物通過0.2 g催化劑(20~40目).使用加熱帶纏繞所有氣體管路,加熱至100 ℃并全程保持,以防止實驗過程中甲苯在裝置管路內壁上冷凝.氣相組成的定量分析由氣相色譜儀(GC-2014,Shimadzu)在線測量,并使用火焰離子化檢測器和熱導檢測器進行檢測.在本研究中,甲苯被完全氧化為H2O和CO2,未檢測到CO或碳氫化合物等副產物.
根據以下公式計算甲苯轉化率(C):
式中:φ0為原料氣體中甲苯的體積分數,φt為對應于具體運行溫度(t)下出口端甲苯的體積分數.
1.4 程序升溫技術
為了探究甲苯在LaCoO3催化劑上的氧化降解情況,在固定床微反應器中進行了原位設計的程序升溫(temperature-programmed,TP)實驗.反應器與OmniStar在線質譜分析儀相連,并連接計算機進行在線測試.在此測試中,0.05 g催化劑樣品先在150 ℃氦氣下預處理1 h,以去除表面弱吸附的H2O和CO2,然后由氦氣吹掃至室溫,保持室溫吸附甲苯,吸附飽和后,用氦氣吹掃整個系統1 h.然后開始進行甲苯的程序升溫表面脫附(temperature-programmed surface desorption of toluene,t-TPD)實驗.通過在氦氣下以10 ℃/min的速率將溫度從室溫升高至500 ℃,同時測定甲苯解吸和CO2生成情況.對于甲苯的程序升溫表面反應(temperature-programmed surface reaction of toluene,t-TPSR)實驗,在程序升溫開始前30 min通入5% O2,并保持此氣氛直至程序升溫結束,其余操作與t-TPD實驗一致.具體實驗步驟如圖2所示.
1.5 原位漫反射傅里葉變換紅外光譜技術
為探究甲苯在催化劑上的吸附-氧化過程,分析研究其反應機理,采用液氮冷卻的MCT探測器,在Thermo Fisher Nicolet iS 10上進行了原位漫反射傅里葉變換紅外光譜(DRIFTS)測試分析.
首先將粉末樣品置于原位DRIFTS反應池內壓平至光滑,通入300 ℃的氦氣進行30 min預處理,以去除表面吸附物質;然后在恒溫循環水槽輔助下迅速降溫至200 ℃并保持,采集并扣除背景值,將甲苯氣流(載氣為氦氣)通入反應池,吸附30 min至吸附曲線不再變化,采集并保存整個過程中的吸附曲線.吸附甲苯后,保持在200 ℃下,通入氦氣吹掃30 min至吸收峰不再發生變化,將20% O2/He混合氣通入原位DRIFTS裝置進行反應后,觀察、采集并保存整個通氧反應過程中的原位DRIFTS變化曲線.
2 結果與討論
2.1 溶膠凝膠形成過程中的溫度影響
對得到的樣品進行XRD測試,結果如圖3所示,所有的樣品都呈現良好的鈣鈦礦結構,對應于LaCoO3(PDF#48-0123).

圖3 不同溶膠凝膠形成溫度制得LaCoO3催化劑的XRD圖
對最終得到的LaCoO3催化劑進行催化氧化甲苯測試,結果如圖4所示.在80 ℃下磁力攪拌加熱得到的LaCoO3催化劑表現出最高的催化活性,在233 ℃時達到90%的甲苯轉化率.但是當磁力攪拌加熱的溫度進一步提高,比如90 ℃和100 ℃,最終得到的LaCoO3催化劑催化氧化甲苯的性能反而略有降低.這可能是因為在水解、縮合形成溶膠以及溶膠聚合成凝膠的過程中,需要足夠的能量以支撐相關反應的進行,只有溫度足夠高時,才能很好保證絡合物的形成[14];但是當溫度過高時,水分蒸發的速度過快也會導致反應物不能得到充分絡合,與此同時,因為溫度過高,聚合反應激烈發生,就會造成膠粒的團聚,最終表現出LaCoO3催化劑催化氧化甲苯活性降低的情況[15].
2.2 鑭鈷鈣鈦礦催化劑煅燒溫度的影響
對不同煅燒溫度得到的催化劑進行XRD測試表征,結果如圖5所示.從圖中可見,500 ℃下煅燒的樣品CAL-500未得到LaCoO3,可能是煅燒溫度不足以形成鈣鈦礦結構.而CAL-600、CAL-700和CAL-800都呈現出良好的鈣鈦礦結構,對應于LaCoO3(PDF#48-0123),除了LaCoO3相外,并未出現La2O3和Co3O4等雜相,表明這些煅燒溫度下得到的樣品都為純相的鑭鈷鈣鈦礦;同時,也觀察到隨著煅燒溫度的升高,樣品的衍射峰明顯變得更加尖銳,而且衍射峰的強度也變得更強,表明伴隨著煅燒溫度的升高,鈣鈦礦相的結晶度也在逐漸增強.

圖5 不同煅燒溫度制得LaCoO3催化劑的XRD圖
對不同煅燒溫度制得樣品進行催化氧化甲苯活性測試,結果如圖6所示,催化氧化甲苯活性由高到低排序為CAL-700>CAL-600≈CAL-800>CAL-500.可以看出在同系列樣品中,CAL-700具有明顯最佳的催化性能,甲苯在CAL-700樣品上90%的轉化率在225 ℃時達到.而未形成鈣鈦礦結構的CAL-500樣品呈現出最低的催化活性,也進一步證明了鈣鈦礦結構具有催化氧化甲苯的優勢.
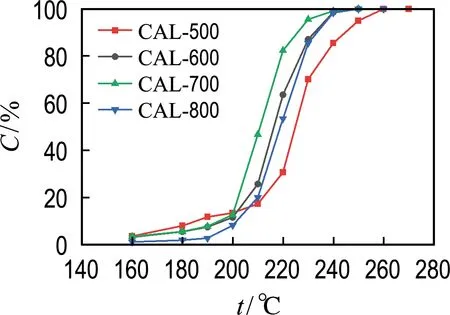
圖6 不同煅燒溫度制得LaCoO3催化劑催化氧化甲苯活性圖
2.3 鑭鈷鈣鈦礦催化劑催化穩定性的研究
對使用同樣的方法、不同批次制備得到的LaCoO3催化劑,在相同條件下進行催化氧化甲苯性能測試,結果如圖7所示.3次制備得到的催化劑催化氧化甲苯性能相當,表明通過溶膠凝膠法,選擇80 ℃形成溶膠凝膠和700 ℃煅燒干凝膠,最終能夠穩定得到LaCoO3催化劑.

圖7 不同批次LaCoO3催化劑反應活性圖
此外,催化劑的工業適用性需考慮其物理和化學穩定性,因此,評價催化劑性能最基本的指標之一是穩定性指標[16].對LaCoO3進行24 h穩定性試驗,如圖8所示,在最初的24 h內,保持反應在240 ℃時進行,LaCoO3催化劑對甲苯的轉化率保持在98%左右.根據研究結果,LaCoO3催化劑催化氧化甲苯的性能在穩定運行24 h后沒有顯著變化,證明了LaCoO3催化劑具有良好的反應穩定性.
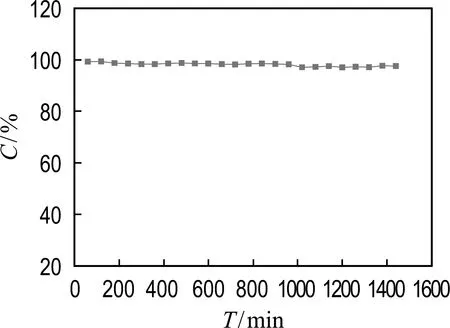
圖8 LaCoO3催化劑的反應穩定性
對24 h穩定運行的LaCoO3催化劑進行XRD測試,以探究穩定性反應后催化劑的結構變化情況,結果如圖9所示.由圖可見,反應后LaCoO3催化劑的結構變化不大,證明了LaCoO3催化劑具有良好的結構穩定性.

圖9 LaCoO3催化劑反應前后的XRD圖
2.4 機理分析
程序升溫實驗結果如圖10所示,在LaCoO3催化劑上,對于吸附的甲苯(m/z=92)來說,與t-TPD反應中的甲苯解吸量相比,t-TPSR中的解吸量降低,表明氣態氧的加入可以促進甲苯的消耗.與此同時,對于產生的CO2(m/z=44)來說,t-TPSR中產生的CO2峰值向低溫方向偏移,表明氣相氧在反應中起著重要作用,可促進甲苯在催化劑上的轉化.

圖10 甲苯程序升溫表面脫附和程序升溫表面反應過程中相關物質變化
采用原位DRIFTS研究甲苯在LaCoO3催化劑上的氧化情況,經過Kubelka-Munk變換后得到吸附曲線隨著時間的變化如圖11(a)所示.由圖可見,隨著甲苯在催化劑表面暴露時間的增加,位于3 072、3 035 cm-1處歸屬于苯環上C—H伸縮振動的吸收峰[17],以及位于2 937、2 880 cm-1處歸屬于甲基上C—H不對稱和對稱伸縮振動的吸收峰在增強[18].1 598、1 494、1 447 cm-1處的吸收峰被歸屬于低溫下典型的芳香環振動[19].逐漸增強的1 418 cm-1吸收峰可歸屬于羧酸基團,表明催化劑表面生成了苯甲酸物種[20].隨著吸附時間的增加,中間體的強度不斷增強,最終達到飽和,表明LaCoO3催化劑本身的氧物種可以參與氧化吸附的甲苯,形成中間產物,但由于活性氧不能持續供應,不足以氧化中間產物.
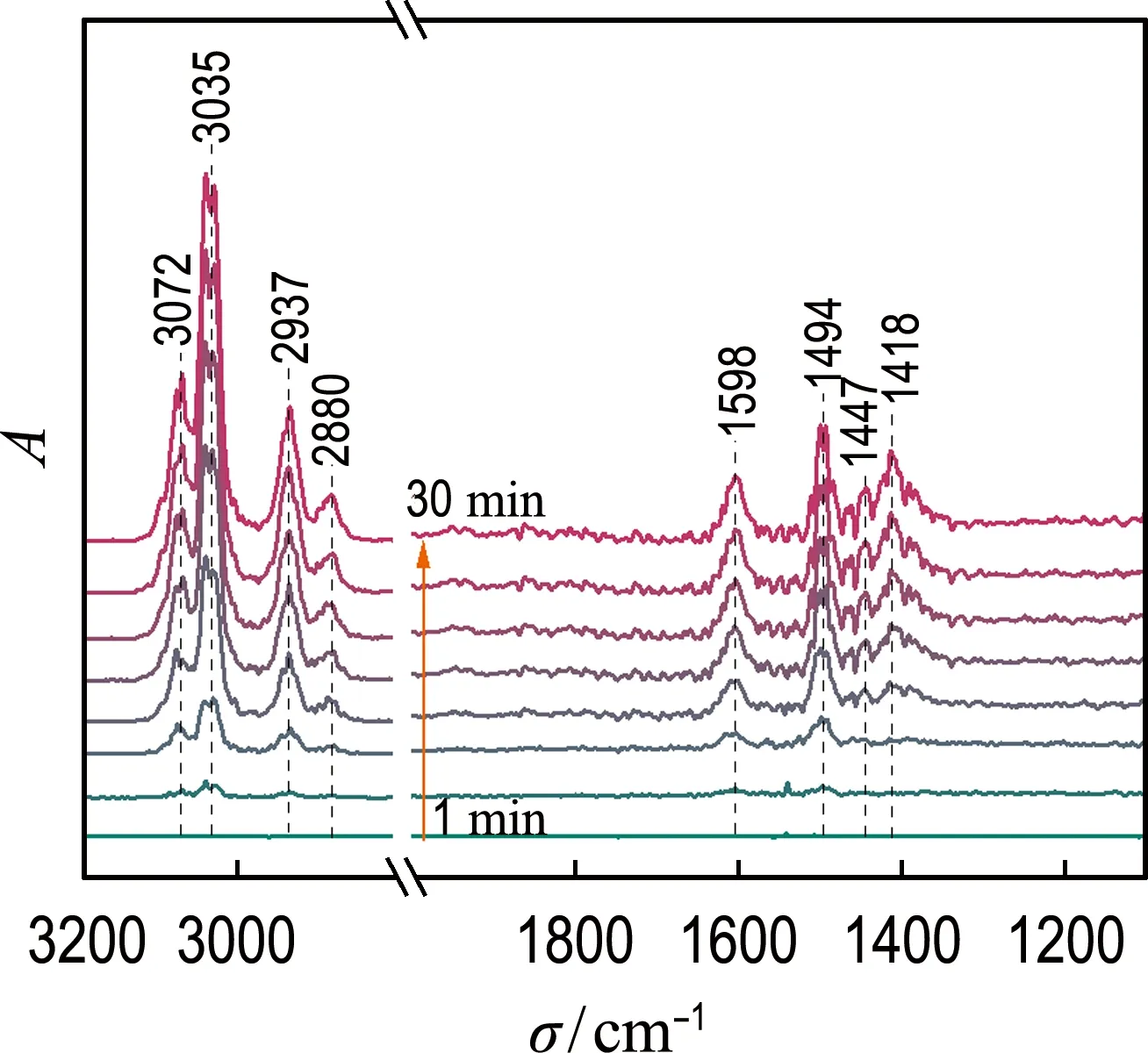
(a) 甲苯吸附
吹掃30 min后,弱吸附的甲苯峰迅速消失,如圖11(b)所示,接著向原位DRIFTS反應池內通入氧氣后,苯甲酸物種的吸收峰先明顯增強后逐漸減弱,說明氣相氧的加入促進了反應的進行.同時,1 539 cm-1處歸屬于小分子羧酸根的吸收峰出現并逐漸增強,說明苯甲酸被逐漸氧化為小分子羧酸鹽[21].結合質譜(圖10)分析結果表明,氣相氧的加入可以有效促進甲苯氧化中間體在LaCoO3催化劑表面的轉化和分解.
3 結 語
本研究使用溶膠凝膠法制備了LaCoO3催化劑,探究了制備溫度對最終得到的催化劑催化氧化甲苯活性的影響.以溶膠凝膠形成溫度80 ℃、煅燒溫度700 ℃的溶膠凝膠法制備得到的LaCoO3催化劑具有最佳的活性,在空速為22 500 mL/(g·h)、甲苯濃度為436 mg/L的條件下,在225 ℃下實現了90%的甲苯轉化率,具有良好的反應穩定性和結構穩定性.結合原位在線質譜和原位漫反射傅里葉變換紅外光譜技術分析機理,表明在LaCoO3催化劑催化氧化甲苯的過程中,催化劑上的氧物種能夠直接參與到甲苯氧化反應中,而氣相氧的加入可以促進甲苯氧化中間體在LaCoO3催化劑表面的轉化和分解.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
