大腦-肋-下頜綜合征一例
2023-10-20 02:21:28胡麗麗孫焱王春祥
放射學實踐 2023年10期
關鍵詞:融合
胡麗麗,孫焱,王春祥
病例資料患兒 女,27 d,G2P2,孕36+6周因“母親妊娠期糖尿病、前置胎盤”擇期行剖宮產出生。因鼻堵、流涕2 d就診。無宮內窒息、生后窒息史,出生體重2.5 kg。孕26周四維B超發現胎兒小下頜。家族史:9歲姐姐及父母均健康。查體:縮頜,懸雍垂至中切牙后1/3裂開,舌根后墜。血常規:WBC13.11×109/L,中性細胞26.1%,淋巴細胞50.2%。
檢查:胸部X線示右側第3-10、左側第3-9后肋裂隙,左側第12肋缺如,脊柱側彎(圖1)。胸部CT容積成像(volume imaging,VR)見雙側多發后肋裂隙及肋橫突關節異常融合(圖2)。胸部CT骨窗見肋椎交界處關節缺失,未發現明顯橫突,肋骨與椎體直接融合(圖3)。頜骨CT VR圖示下頜骨小伴輕度后縮(圖4)。結合臨床及影像診斷為大腦-肋-下頜綜合征。
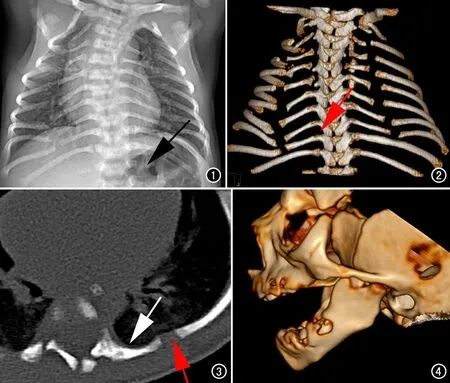
圖1 胸部X線正位片示右側第3-10后肋、左側第3-9后肋裂隙,左側第12肋缺如(箭),脊柱側彎。 圖2 胸部CT VR示雙側多發后肋裂隙及肋橫突關節異常融合(箭)。 圖3 胸部CT骨窗圖示肋椎交界處關節缺失,未發現明顯橫突,肋骨與椎體直接融合(白箭),后肋裂隙邊緣皮質良好(紅箭)。 圖4 頜骨CT VR示下頜骨小伴輕度后縮。
討論大腦-肋-下頜綜合征(cerebro-costo-mandibular syndrome,CCMS)罕見,是一種以后肋裂隙和皮羅綜合征(小下頜、舌根后墜、腭裂)為特征的多發畸形,最早由Smith等[1]于1966年提出。本病是常染色體顯性遺傳病,研究報道顯示CCMS是由SNRPB基因雜和突變導致[2]。目前已有超過80例報道,本例為首例中國大陸報道。CCMS主要顱面特征是小下頜,常伴發腭裂、進食和呼吸困難。其他表現有神經發育遲緩、脊柱側彎、后鼻孔閉鎖、喉氣管畸形、心臟、腎臟和聽力異常[3]。Nagasawa等[4]據嚴重程度將CCMS分為3型:致死型,患兒生后1個月內死亡;嚴重型,患兒生后1~12個月內死亡;輕型,患兒生存時間超過1年。
本病據典型臨床及X線表現即可確診。特征性影像學表現為多發后肋裂隙、肋骨數量減少、肋橫突關節異常融合。其中多發后肋裂隙是診斷性表現[5,6]。本例患者影像表現比較典型。組織學上肋骨裂隙是肋骨后部被纖維血管組織所取代,這些組織最終可能會發生鈣化[7,8]。后肋裂隙僅見于另外2種疾病[9],透明椎骨發育障礙(diaphanospondylodysostosis, DSD)和IIg型先天性糖基化障礙。DSD椎體無骨化或骨化嚴重延遲、腎母細胞瘤病伴囊性腎,無小頜畸形、腭裂。IIg型先天性糖基化障礙可通過肋骨融合、脊椎分節不良以及其他表現如腦結構異常、畸形足、小耳畸形和黃斑病變來鑒別。后肋裂隙的其他鑒別診斷包括多發肋骨骨折,尤其是心肺復蘇后患兒可通過復查胸部X線片觀察有無骨痂來鑒別[10]。皮羅綜合征也可表現為小頜畸形但無后肋裂隙。總之,CCMS的影像表現具有特征性,結合臨床及典型影像表現即可確診。
猜你喜歡
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
數學年刊A輯(中文版)(2022年4期)2022-02-16 08:17:34
今日農業(2021年19期)2022-01-12 06:16:36
中老年保健(2021年11期)2021-08-22 03:15:44
無線電通信技術(2021年4期)2021-07-13 08:58:28
無線電通信技術(2021年3期)2021-06-08 03:33:48
中學生數理化(高中版.高考數學)(2021年1期)2021-03-19 08:28:38
無線電工程(2020年11期)2020-10-29 01:25:46
現代出版(2020年3期)2020-06-20 07:10:34
福利中國(2015年4期)2015-01-03 08:03:38
