葉黃素納米混懸劑制備及其體內藥動學研究
2023-10-30 06:11:46陳永順蔣建平梁建梅賈曉棟
中成藥 2023年10期
陳永順,李 靜,蔣建平,梁建梅,楊 斌,賈曉棟
(1.商丘醫學高等專科學校,河南 商丘 476000; 2.廣西醫科大學,廣西 南寧 530021)
葉黃素是一種含有2 個紫羅酮環的天然類胡蘿卜素,廣泛存在于胡蘿卜、萬壽菊等植物中,具有抗氧化、預防心腦血管疾病、保護腦功能、預防白內障等活性[1-2],通過免疫調節、抑制腫瘤細胞生長等機制對皮膚癌、食管癌、前列腺癌、胃癌等有較強活性[2-3],長期毒性研究顯示,該成分基本無不良反應,安全性較高[4]。但葉黃素溶解度僅為(6.69±0.10) μg/mL,溶出度較低; 表觀油水分配系數為1.03[5],具有一定脂溶性; 相對生物利用度只有2% ~9.4%[6],目前已有脂質體[7]、固體分散體[3]、納米晶體[8]等相關報道。
前期報道,納米混懸劑通過降低藥物粒徑來實現增加溶解度、促進溶出及體內吸收等作用[9-12],黃雅蘭等[9]以十六烷基三甲基氯化鈉為乳化劑制備了葉黃素納米晶體,但它有一定毒性[13],需加入大量凍干保護劑制成凍干粉,從而增加了輔料種類及用量。白蛋白是一種天然乳化劑,安全性高,本身還可作為凍干保護劑[14],可減少輔料用量,故本實驗以牛血清白蛋白為穩定劑制備葉黃素納米混懸劑[15-17],并考察其體內藥動學,以期為相關新型制劑開發提供新的思路。
1 材料
Agilent 1100 型高效液相色譜儀(美國Agilent公司); CD-X15 型超聲儀(深圳市亦為實業有限公司); JB-1 型磁力攪拌器(上海葉拓科技有限公司); MSE125P-CE 型電子天平(德國Sartorius 公司); YSB-BCF12 型超低溫冰箱(佛山市雅紳寶制冷設備制造有限公司); RC-6D 型溶出儀(天津創興電子設備制造股份有限公司); BMH 型真空凍干機(北京博勱設備有限公司); Master-sizer 型粒度分析儀(英國馬爾文儀器有限公司); HAC-I 型氮吹儀(北京萊寧德銳科技有限公司)。
葉黃素對照品(批號200510,純度92%,上海如吉生物科技有限公司); 葉黃素原料藥(批號20200812,純度90%,成都彼樣生物科技有限公司); 麥角甾醇對照品 (批號20220522,純度98%,成都嘉葉生物科技有限公司)。吐溫-80 (批號20201212,天津紅太陽化工有限公司); 牛血清白蛋白(BSA,批號20201118,上海藍季生物科技有限公司); 十二烷基硫酸鈉(SDS,批號191006,國藥集團化學試劑有限公司); 甘露醇 (批號190915,湖南九典制藥股份有限公司)。
SD 大鼠,雌雄兼具,體質量 (220±20) g,購自河南省實驗動物中心,動物生產許可證號SCXK (豫) 2020-0012。
2 方法與結果
2.1 葉黃素含量測定
2.1.1 色譜條件 Hypersil ODS C18色譜柱(250 mm×4.6 mm,5 μm); 流動相甲醇-乙腈-二氯甲烷(55 ∶ 25 ∶ 20); 體積流量1.0 mL/min; 柱溫35 ℃; 檢測波長446 nm; 進樣量10 μL。
2.1.2 供試品溶液制備 取納米混懸劑1 mL,置于50 mL 量瓶中,加入40 mL 甲醇超聲處理5 min,甲醇稀釋至刻度,0.45 μm 微孔濾膜過濾,精密量取2 mL 至10 mL 量瓶中,流動相稀釋至刻度,即得。
2.1.3 對照品溶液制備及線性關系考察 取葉黃素對照品適量,甲醇制成質量濃度為200 μg/mL的貯備液,流動相依次稀釋至5、2.5、1、0.5、0.1、0.05 μg/mL,即得。以對照品質量濃度為橫坐標(X),峰面積為縱坐標(Y) 進行回歸,得方程為Y=715.84X+126.5 (r=0.999 8),在0.05 ~5 μg/mL 范圍內線性關系良好。
2.1.4 方法學考察 取0.05、1、5 μg/mL 對照品溶液適量,在“2.1.1” 項色譜條件下進樣測定6次,測得葉黃素含量RSD 分別為0.22%、0.60%、0.43%,表明儀器精密度良好。取納米混懸劑適量,按“2.1.2” 項下方法平行制備6 份供試品溶液,在“2.1.1” 項色譜條件下進樣測定,測得葉黃素含量RSD 為1.70%,表明該方法重復性良好。取供試品溶液適量,室溫下于0、3、6、9、12、24 h 在“2.1.1” 項色譜條件下進樣測定,測得葉黃素含量RSD 為0.48%,表明溶液在24 h 內穩定性良好。取9 份納米混懸劑,每份0.5 mL,置于50 mL 量瓶中,分為低、中、高3 組,分別加入對照品貯備液1、2、3 mL,按“2.1.2” 項下方法制備供試品溶液,在“2.1.1” 項色譜條件下進樣測定,測得葉黃素平均加樣回收率分別為100.63%、99.47%、101.08%,RSD 分別為0.36%、0.89%、0.77%。
2.2 納米混懸劑制備 參考文獻[17-18] 報道,取葉黃素原料藥30 mg,溶于5 mL 四氫呋喃中,作為有機相; 配制一定濃度含吐溫-80、牛血清白蛋白的水溶液50 mL,45 ℃水浴加熱,作為水相,將有機相緩慢滴入水相中,800 r/min 磁力攪拌40 min,置于均質機中循環均質,0.45 μm 微孔濾膜過濾,蒸餾水補足至50 mL,置于冰水混合物中降溫,即得。
2.3 粒徑、PDI、Zeta 電位測定 取納米混懸劑0.5 mL,蒸餾水稀釋50 倍,渦旋10 s 混勻,在粒度分析儀上測定,平行3 次,取平均值。
2.4 處方優化 采用單因素試驗。
2.4.1 白蛋白濃度 固定葉黃素用量30 mg,吐溫-80 濃度0.5%,均質壓力80 MPa,均質次數6次,考察白蛋白濃度對粒徑、PDI 的影響,結果見圖1。由此可知,處方中僅有吐溫-80 時粒徑接近1 000 nm,PDI 大于0.6; 隨著白蛋白濃度增加粒徑先降低后趨穩,而PDI 先降低后升高,為1.5%時兩者最低。最終確定,白蛋白濃度為1.5%。

圖1 白蛋白濃度對粒徑、PDI 的影響(n=3)Fig.1 Effects of albumin concentration on particle size and PDI (n=3)
2.4.2 吐溫-80 濃度 固定葉黃素用量30 mg,白蛋白濃度1.5%,均質壓力80 MPa,均質次數6次,考察吐溫-80 濃度對粒徑、PDI 的影響,結果見圖2。由此可知,白蛋白濃度為1.5% (不加吐溫-80) 時粒徑約為1 500 nm,PDI 大于0.7; 隨著吐溫-80 濃度增加粒徑、PDI 均先降低后趨穩,大于0.75%時兩者變化不明顯。最終確定,吐溫-80濃度為0.75%。
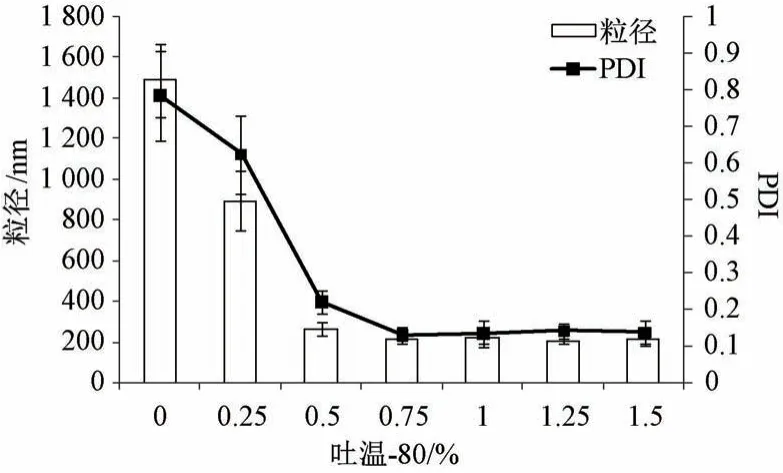
圖2 吐溫-80 濃度對粒徑、PDI 的影響(n=3)Fig.2 Effects of Tween-80 concentration on particle size and PDI (n=3)
2.4.3 均質壓力 固定葉黃素用量30 mg,白蛋白濃度1.5%,吐溫-80 濃度0.75%,均質次數6次,考察均質壓力對粒徑、PDI 的影響,結果見圖3。由此可知,隨著均質壓力增加粒徑、PDI 均先降低后升高,可能是因為壓力較小時無法提供足夠能量形成較小粒徑的納米粒,而過大時可能會導致體系溫度較高,破壞納米粒保護層[18-19]并發生融合、聚集,導致兩者變大。最終確定,均質壓力為80 MPa。

圖3 均質壓力對粒徑、PDI 的影響(n=3)Fig.3 Effects of homogenization pressure on particle size and PDI (n=3)
2.4.4 均質次數 固定葉黃素用量30 mg,白蛋白濃度1.5%,吐溫-80 濃度0.75%,均質壓力80 MPa,考察均質次數對粒徑、PDI 的影響,結果見圖4。由此可知,隨著均質次數增加粒徑、PDI均先降低后升高,可能是因為次數過多時體系溫度急劇升高,納米粒之間容易發生融合、聚集,粒徑分布不均勻,導致兩者變大,并且為8 次時均降低。最終確定,均質次數為8 次。

圖4 均質次數對粒徑、PDI 的影響(n=3)Fig.4 Effects of homogenization frequency on particle size and PDI (n=3)
2.5 驗證試驗 “2.4” 項下結果顯示,最優處方為葉黃素用量30 mg,白蛋白濃度1.5%,吐溫-80 濃度0.75%,均質壓力80 MPa,均質次數8 次,平均粒徑為(208.71±9.26) nm,PDI 為0.114±0.017,Zeta 電位為- (23.15±1.60) mV,見圖5~6。
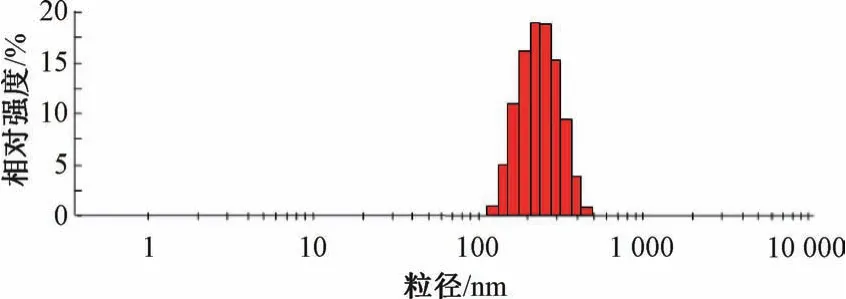
圖5 葉黃素納米混懸劑粒徑分布Fig.5 Particle size distribution of lutein nanosuspensions

圖6 葉黃素納米混懸劑Zeta 電位Fig.6 Zeta potential of lutein nanosuspensions
2.6 形態觀察 取納米混懸劑0.5 mL,蒸餾水稀釋50 倍,混勻,滴于銅膠帶上,室溫晾干,噴金1 min,再按處方工藝制備空白樣品(不含葉黃素,其余輔料比例同納米混懸劑),同法測定,在掃描電鏡(放大15 000 倍) 下觀察,結果見圖7。由此可知,納米混懸劑基本呈球形,而空白樣品(不含葉黃素) 中未發現納米粒。

圖7 葉黃素納米混懸劑(A)、空白樣品(B) 掃描電鏡圖Fig.7 Scanning electron microscopy images of lutein nanosuspensions (A) and blank sample (B)
2.7 凍干粉制備及表征
2.7.1 制備工藝 取納米混懸劑適量,加入一定質量分數甘露醇,渦旋30 s 至澄清,分裝至西林瓶中(每份均3 mL),密封置于-30 ℃冰箱中預凍2 d后立即轉移至冷凍干燥機(冷阱溫度-55 ℃)中,當真空度達到0.01 kPa 時凍干2 d,取出,即得,蒸餾水復溶,考察甘露醇用量對粒徑、PDI 的影響,結果見圖8。由此可知,不添加甘露醇時凍干粉復溶后粒徑為(252.67±18.13) nm,PDI 為(0.132±0.013),可能與白蛋白本身是一種優良凍干保護劑有關,但其外觀很差; 隨著甘露醇用量增加凍干粉外觀逐漸達飽滿狀態,但超過2%時粒徑有增加趨勢,超過3%時PDI 也開始變大。最終確定,以1.5%甘露醇為凍干保護劑,此時凍干粉外觀飽滿,復溶后粒徑、PDI 較小。

圖8 甘露醇用量對粒徑、PDI 的影響(n=3)Fig.8 Effects of mannitol consumption on particle size and PDI (n=3)
2.7.2 晶型分析 設定條件為掃描范圍3°~45°,電壓40 kV,掃描速度6°/min,Cu-Kα 靶。取原料藥、空白輔料(不含藥物,輔料比例同納米混懸劑凍干粉)、物理混合物、新鮮納米混懸劑凍干粉、放置6 個月納米混懸劑凍干粉(溫度30 ℃,相對濕度65%) 適量,進行X 射線粉末衍射(XRPD) 分析,結果見圖9。由此可知,原料藥在20.3°、28.4°、40.2°處出現特征晶型峰[4]; 由于輔料掩蔽作用,物理混合物圖譜中僅發現原料藥在40.2°處的晶型峰,表明它以晶型狀態存在; 新鮮納米混懸劑凍干粉圖譜中未發現原料藥晶型峰,表明它可能轉變為無定形狀態; 放置6 個月納米混懸劑凍干粉圖譜中仍發現原料藥晶型峰,表明其穩定性良好。
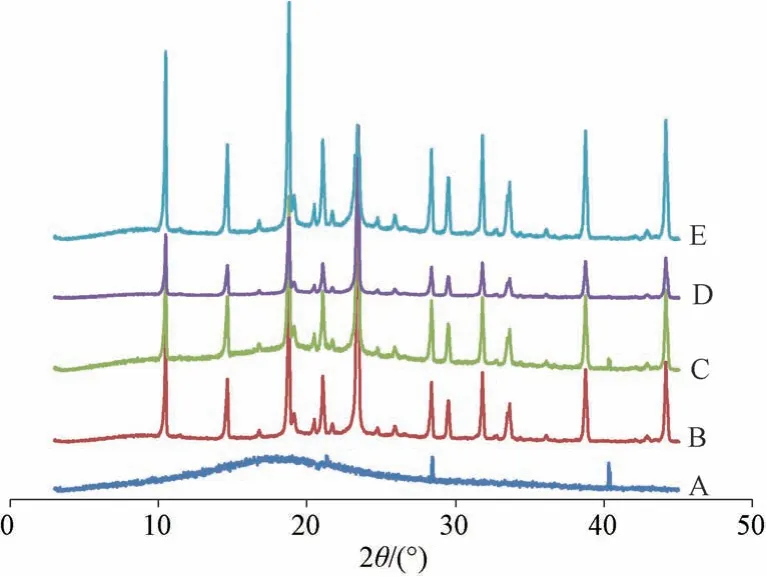
圖9 各樣品XRPD 圖Fig.9 XRPD patterns for various samples
2.8 溶解度測定 取過量原料藥、物理混合物、納米混懸劑凍干粉至燒杯中,加入10 mL 蒸餾水,在25 ℃水浴中800 r/min 磁力攪拌72 h,取混懸液,0.45 μm 微孔濾膜過濾,取續濾液,測定溶解度。結果,原料藥、物理混合物溶解度分別為(6.69±0.10)、(6.94±0.11) μg/mL,而納米混懸劑凍干粉達(308.57±2.02) μg/mL,即與原料藥相比增加至46.12 倍。
2.9 體外溶出研究 由于葉黃素在1.5%SDS 溶液中的溶解度為46.47 μg/mL,故10 mg 該成分在1 000 mL 1.5%SDS 溶液中即可達漏槽條件。取納米混懸劑凍干粉(含10 mg 葉黃素) 適量,加入1.5%SDS 溶液5 mL,置于透析袋中(截留分子量8 000 ~12 000 Da),扎緊,設定釋藥介質為1 000 mL 1.5% SDS 溶液(溫度37 ℃),轉速為75 r/min,于0、5、15、30、45、60、90、120、240、360、480 min 各取樣5 mL,并補加5 mL 空白介質,0.45 μm 微孔濾膜過濾,取續濾液,測定溶出度,同法測定原料藥、物理混合物,結果見圖10。由此可知,原料藥在漏槽條件下480 min 內溶出度僅為19.80%,可能與其粒度較大、疏水性較強有關[19],而納米混懸劑凍干粉在360 min 內累積溶出度即達95%。

圖10 各樣品體外溶出曲線(n=6)Fig.10 In vitro dissolution curves for various samples (n=6)
2.10 穩定性研究 取納米混懸劑凍干粉適量,蒸餾水復溶,測定離心前粒徑,再取2 mL 至離心管中,1 500 r/min 離心30 min,取上層混懸液測定離心后粒徑,計算穩定系數[20],公式為穩定系數=離心后粒徑/離心前粒徑(越接近1,樣品越穩定),然后置于溫度30 ℃、相對濕度65% 的恒溫恒濕箱中,于0、3、6、9、15、30、45、60、90 d 同法測定,結果見圖11。由此可知,納米混懸劑凍干粉放置90 d 后穩定系數仍大于0.95,表明其穩定性良好。

圖11 葉黃素納米混懸劑不同時間點穩定系數(n=3)Fig.11 Stability coefficients of lutein nanosuspensions at different time points (n=3)
2.11 體內藥動學研究
2.11.1 分組、給藥與采血 取原料藥、物理混合物、納米混懸劑凍干粉適量,0.5% CMC-Na 溶液制成藥液(以葉黃素計,質量濃度為4 mg/mL)。18 只大鼠隨機分為3 組,每組6 只,按30 mg/kg劑量灌胃給藥,于0.5、1、1.5、2、2.5、3、4、6、12、18 h 眼眶靜脈叢取血各0.25 mL,置于肝素浸潤離心管中,密封,渦旋5 s 混勻,3 000 r/min離心2 min,取出血漿,冷凍保存。
2.11.2 血漿樣品處理 參考文獻[21] 報道,取麥角甾醇對照品適量,乙腈稀釋至886 ng/mL,作為內標溶液,取50 μL,加到100 μL 血漿樣品中,渦旋混勻1 min,加入1 mL 氯仿,渦旋3 min,4 ℃、10 000 r/min 離心10 min,收集氯仿層,40 ℃氮吹儀吹干,100 μL 乙腈復溶,10 000 r/min離心10 min。
2.11.3 線性關系考察 取葉黃素對照品適量,乙腈制成2 080、1 040、520、260、104、52 ng/mL溶液,分別取100 μL 至離心管中,40 ℃氮吹儀吹干,殘渣中加入100 μL 空白血漿,渦旋3 min,按“2.11.2” 項下方法處理,即得質量濃度分別為2 080、1 040、520、260、104、52 ng/mL 的血漿對照品溶液(含內標)。以對照品質量濃度為橫坐標(X),葉黃素、麥角甾醇峰面積比值為縱坐標(Y) 進行回歸,得方程為Y=0.001 9X-0.236 7(r=0.995 4),在52 ~2 080 ng/mL 范圍內線性關系良好。
2.11.4 方法學考察 取52、520、2 080 ng/mL血漿對照品溶液適量,同一天內在“2.1.1” 項色譜條件下進樣測定6 次,測得葉黃素、麥角甾醇峰面積比值RSD 分別為6.05%、5.26%、5.06%,表明該方法日內精密度良好; 同法連續測定6 d,每天1 次,測得兩者峰面積比值RSD 分別為9.43%、6.00%、7.52%,表明該方法日間精密度良好。取給藥0.5 h 后血漿樣品適量,于0、3、6、9、12、24 h 在“2.1.1” 項色譜條件下進樣測定,測得葉黃素、麥角甾醇峰面積比值RSD 為7.44%,表明樣品在24 h 內穩定性良好。取上述3 種質量濃度血漿對照品溶液適量,按“2.11.2” 項下方法處理,在“2.1.1” 項色譜條件下進樣測定,測得葉黃素平均加樣回收率分別為93.67%、96.08%、93.82%,RSD 分別為6.20%、7.35%、4.69%。
2.11.5 結果分析 血藥濃度-時間曲線見圖12,主要藥動學參數見表1。由此可知,原料藥、物理混合物相關數值比較,差異無統計學意義 (P>0.05),表明輔料對原料藥口服吸收基本無影響;與原料藥、物理混合物比較,納米混懸劑tmax縮短(P<0.05),Cmax、AUC0~t、AUC0~∞升高 (P<0.01),相對生物利用度相較于原料藥增加至3.71倍,但t1/2無明顯變化(P>0.05)。
表1 葉黃素主要藥動學參數(±s,n=6)Tab.1 Main pharmacokinetic parameters for lutein (±s,n=6)
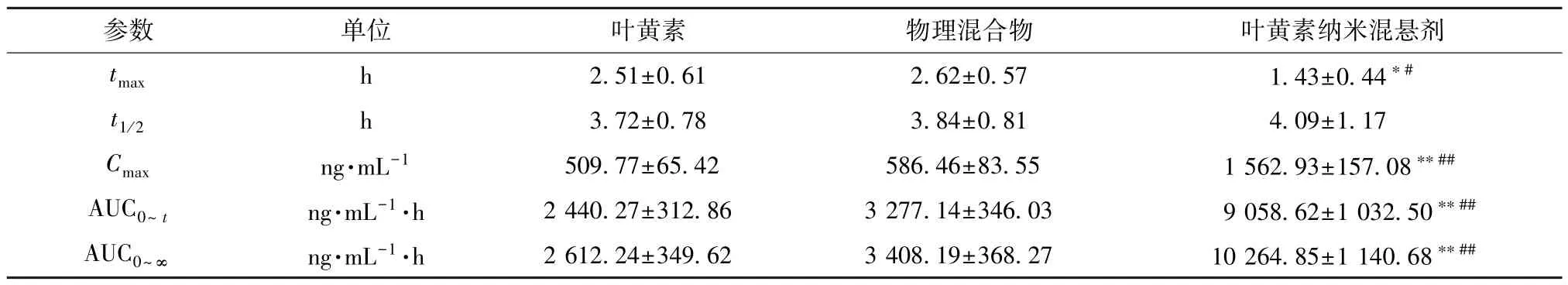
表1 葉黃素主要藥動學參數(±s,n=6)Tab.1 Main pharmacokinetic parameters for lutein (±s,n=6)
注: 與葉黃素比較,*P<0.05,**P<0.01; 與物理混合物比較,#P<0.05,##P<0.01。
參數單位葉黃素物理混合物葉黃素納米混懸劑tmaxh2.51±0.612.62±0.571.43±0.44*#t1/2h3.72±0.783.84±0.814.09±1.17 Cmaxng·mL-1509.77±65.42586.46±83.551 562.93±157.08**##AUC0~tng·mL-1·h2 440.27±312.863 277.14±346.039 058.62±1 032.50**##AUC0~∞ng·mL-1·h2 612.24±349.623 408.19±368.2710 264.85±1 140.68**##
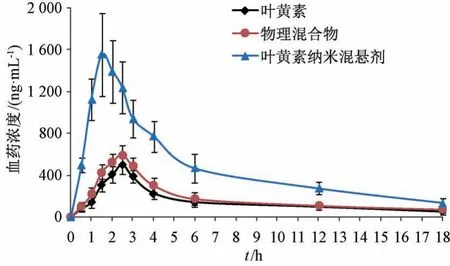
圖12 葉黃素血藥濃度-時間曲線(n=6)Fig.12 Plasma concentration-time curves for lutein (n=6)
3 討論與結論
白蛋白吸附在納米粒表面并發生交聯后形成界面膜,伸向水相的長鏈提供了巨大的空間位阻,從而起到穩定納米粒的作用[22]。為了減小納米混懸劑粒徑,本實驗在處方中引入非離子表面活性劑吐溫-80[23]。另外,處方中白蛋白除了作為穩定劑外,本身也是一種凍干保護劑,只需1.5%甘露醇即可制得外觀飽滿的凍干粉,大大降低了輔料用量。
白蛋白在均質條件下可形成納米粒[12-13,24],但本實驗按處方工藝制備的葉黃素納米混懸劑空白樣品中并未發現納米粒,可能是由于吐溫-80 通過氫鍵結合到白蛋白疏水區,增強了后者穩定性,從而阻止其變性形成納米粒[25-26]。另外,葉黃素納米混懸劑中原料藥晶型發生了變化,可能是除去有機溶劑時后者逐漸析出,此時穩定劑的空間穩定作用、纏繞作用、靜電作用等抑制了結晶形成,最終使其以無定形狀態存在[9]。
體內藥動學研究結果顯示,葉黃素納米混懸劑tmax顯著縮短,可能與其前期釋藥速率較快有關;Cmax、相對生物利用度分別增加至3.06、3.71 倍,可能是由于納米混懸劑可大大提高原料藥溶解度、累積溶出度,解決了吸收瓶頸: 納米混懸劑可使原料藥比表面積更大,增加與胃腸道的接觸面,有利于吸收[23]。另外,葉黃素屬于生物藥劑學Ⅳ藥物[5],盡管納米混懸劑有效改善了其水溶性、溶出度,但可能對脂溶性的影響有限,故會限制其生物利用度的提高程度。
綜上所述,本實驗完成了葉黃素納米混懸劑的制備及其體內藥動學的考察,可為后續該制劑相關劑型(片劑或膠囊劑) 開發、藥效學評價等方面奠定堅實基礎。
