雙模板分子印跡材料固相萃取-高效液相色譜法聯用測定果蔬中三嗪類農藥
2023-11-07 04:15:34劉建輝鞏碧釧胡秋輝蘇安祥謝旻皓楊文建
食品科學 2023年20期
關鍵詞:檢測
劉建輝,鞏碧釧,胡秋輝,蘇安祥,徐 輝,謝旻皓,楊文建
(南京財經大學食品科學與工程學院,江蘇省現代糧食流通與安全協同創新中心,江蘇省食用菌保鮮與深加工工程研究中心,江蘇 南京 210023)
三嗪類除草劑因其高效、廣譜、廉價等作用特點,在全球范圍內普遍使用,且廣泛應用于果蔬等農作物的生產中[1]。然而,該類除草劑的立體化學反應穩定性高,環境持久性長,在施用過程中可遷移至水體、土壤和農作物,進而進入至人體,對人體產生致癌和內分泌干擾等不可逆的副作用,嚴重危害人類健康[2-3]。此外,三嗪類農藥可產生多種降解產物,如脫乙基莠去津、脫異丙基莠去津和羥基化莠去津等[4],其毒性類似或更甚于原物質[5]。目前,已在土壤、地下水、飲用水、農作物、水生動物等中均能檢測到三嗪類除草劑及其降解物殘留[6-7]。三嗪可以吸附到水果或蔬菜的表面/表皮,并最終滲透到水果或蔬菜的果肉中[8]。
針對三嗪類除草劑極易在環境與食物中殘留從而對生物體帶來嚴重危害,許多國家都對食品中的殘留量進行了限制,例如歐盟已經停止了該類除草劑在農業生產中的應用,美國環保署將其列入了優先控制的污染物名單[9]。我國最新GB 2763—2021《食品中農藥最大殘留限量》對蔬菜、水果、谷類、油料和油脂中三嗪類除草劑的最大殘留限量為0.01~0.5 mg/kg[10]。但因其極易殘留的特點,仍需建立高效的檢測方法以監測其在環境與食品中的殘留。然而,農作物樣品基質復雜,且三嗪類農藥代謝物通常以結合狀態存在,因此在檢測前的樣品前處理過程比較繁瑣。在眾多已報道的前處理方法中,固相萃取技術應用最廣。但通用型的固相萃取柱如C鍵合硅膠等非選擇性的吸附溶劑,選擇性和特異性差,難以用于復雜基體中微量物質的吸附萃取[11]。因此,開發一種凈化富集能力強的前處理技術,對于檢測復雜樣品中三嗪類化合物具有重要意義。
分子印跡聚合物(molecularly imprinted polymers,MIPs)對目標分子(稱為“模板”)具有結合性能高、特異性強和穩定性好等優點,特別適用于痕量分析[12]。孔光輝等[13]以莠去津為模板建立了分子印跡固相萃取方法,可萃取并檢測煙葉中的三嗪類除草劑。Zhou Tianyu等[14]制備了阿特拉津MIPs,其工藝簡單、成本低,并通過結合MIPs固相萃取與高效液相色譜-質譜(high performance liquid chromatograph-mass spectrometry,HPLC-MS)技術,成功吸附并檢測了茶葉樣品中的三嗪類化合物。但目前多數MIPs只停留在針對單一模板的印跡聚合物的制備,識別位點單一,不能對多種三嗪類目標分子產生特異性作用[15]。而使用兩種或多種結構類似物作為模板,多模板分子印跡可以擴展單模板分子印跡的應用,以實現同時識別、提取和分離一種以上分析物[16-17]。
本研究團隊前期制備了以滅蠅胺為虛擬模板的MIPs,提高了三嗪類農藥的檢測效率和精度[18]。由于三嗪類除草劑的特性官能團為三嗪環,除選取滅蠅胺外,也可聯用同樣結構相似的三聚氰胺作為替代的雙虛擬模板分子[19]。為進一步增加識別與吸附位點,提高對三嗪類化合物的吸附效率,本研究擬聯用滅蠅胺和三聚氰胺,開發雙虛擬模板分子印跡聚合物,并對材料進行優化與吸附性能表征,聯合應用HPLC法檢測蘋果、黃瓜、玉米等果蔬中的三嗪類農藥殘留。
1 材料與方法
1.1 材料與試劑
蘋果、黃瓜、玉米等樣品 江蘇省南京市蘇果超市;農藥標準品滅蠅胺、三聚氰胺、阿特拉津、撲草凈、西草凈、莠滅凈、吡蟲啉(純度均大于98%)以及甲基丙烯酸(methacrylic acid,MAA)、乙二醇二甲基丙烯酸酯(ethylene dimethacrylate,EGDMA)、三羥甲基丙烷三甲基丙烯酸酯(trimethylpropane trimethacrylate,TRIM)、偶氮二異丁腈(azobisisobutyronitrile,AIBN)(均為分析純)、乙腈(色譜純)美國Sigma公司;冰乙酸、甲醇、三氯甲烷(均為分析純)上海阿拉丁生化科技股份有限公司。
1.2 儀器與設備
Nicolet 6700傅里葉變換紅外光譜儀 美國Thermo Fisher公司;DZ-3BCII真空干燥箱 上海奧析科學儀器有限公司;ZNCL-G15型智能磁力攪拌器 河南愛博特科技發展有限公司;Allegra 64R離心機 美國貝克曼庫爾特公司;固相萃取裝置 美國Supelco公司;SU8010掃描電鏡 日本Hitachi公司;1260系列HPLC儀 美國Agilent公司;HSC-24A氮吹儀 南京科捷分析儀器有限公司。
1.3 方法
1.3.1 雙模版MIPs制備及優化
稱取0.6 mmol滅蠅胺、0.4 mmol三聚氰胺和4 mmol MAA,向其中加入15 mL乙腈-水溶液(3∶1,V/V)進行混合,超聲30 min以使其預聚合。之后向其中加入與功能單體MAA等量的交聯劑TRIM,以及30 mg AIBN再進行超聲5 min,之后為排除其中氧氣,置于氮吹儀中通入氮氣5 min。封閉混合液后再將其置于60 ℃水浴條件下持續24 h。反應結束之后獲得塊狀雙模版分子聚合物,采用甲醇-乙酸(4∶1,V/V)溶液將研磨后的模板分子洗脫,用HPLC儀檢測直至提取液中無模板分子存在。所得印跡聚合物分別用甲醇和水沖洗3 次以上,于60 ℃真空干燥。非印跡聚合物(non-imprinted polymers,NIPs)作為對照,其制樣方法與MIPs相同,區別在于無須添加兩種模板分子。同時,為使MIPs的吸附量達到最佳,選取不同的制孔劑(乙腈與三氯甲烷)、交聯劑(EGDMA和TRIM)以測定MIPs的吸附量,并且對雙模板中滅蠅胺與三聚氰胺的含量進行優化。
1.3.2 雙模板分子印跡聚合物表征
1.3.21 紅外光譜表征
將干燥溴化鉀粉末充分研磨,用壓片機壓片,測定其紅外光譜以調整基線。稱取5 mg干燥的MIPs、NIPs和滅蠅胺、三聚氰胺標品以及溴化鉀粉末,研磨均勻并壓成薄片,于4000~500 cm-1范圍分別掃描,以表征其紅外光譜[20]。
1.3.22 掃描電鏡表征
將導電膠涂抹并分散在待測試樣上,并對試樣再鍍金處理,在掃描電鏡下觀察試樣結構。
1.3.3 雙模板分子印跡聚合物吸附性能
1.3.31 動力學吸附實驗
分別稱量10 mg MIPs與NIPs,溶于80 mg/L的撲草凈標準溶液中并于25 ℃水浴中充分攪拌。于不同時間點(15、30、60、90、120、150、180、210、240 min)進行取樣,離心后取上清液,過0.22 μm濾膜,應用HPLC檢測其含量,并利用式(1)計算試樣對撲草凈溶液的吸附量(Q):
式中:Q為平衡狀態時聚合物對目標分子的吸附量/(μ g/g);C0為標準農藥溶液的初始質量濃度/(m g/L);C為標準農藥溶液的平衡質量濃度/(mg/L);V為所加農藥溶液的體積/mL;M為MIPs或NIPs的添加量/mg。
1.3.32 靜態吸附實驗及Scatchard模型擬合
準確稱量雙模版MIPs和非印跡聚合物NIPs各10 mg,加入1 mL不同質量濃度(25、50、75、100、125、150、175、200 mg/L)撲草凈標準溶液。在25 ℃恒溫環境下振蕩2 h,5000 r/min離心10 min后采用HPLC方法測定上清液中目標分析物的濃度。利用式(1)計算MIPs、NIPs對撲草凈溶液的吸附量。
按照式(2)繪制Scatchard標準曲線,據此得出聚合物的吸附常數Kd與最大表觀吸附量Qmax。
式中:Q為聚合物對目標分子的吸附量/(μg/g);C為目標物質的平衡濃度/(mg/L);Qmax為吸附位點的最大表觀結合量/(μg/g);Kd為吸附位點的解離平衡常數/(μg/L)。
1.3.33 選擇性吸附實驗
將10 mg MIPs分別與1 mL的撲草凈、阿特拉津、莠滅凈、西草凈以及吡蟲啉農藥標準溶液(80 mg/L)混合均勻,并置于室溫混合2 h,利用HPLC測定上層清液的濃度。同時作為對照,按同樣方法測定MIPs對滅蠅胺和三聚氰胺的吸附能力。計算雙模版MIPs的印跡因子和選擇性指數以評判其特異性吸附能力[21]。
1.3.4 HPLC檢測條件
待測液制備好后,HPLC儀裝備C18色譜柱(250 mm×4.6 mm,5 μm),上樣量為10 μL,以甲醇-水(7∶3,V/V)作為流動相,流速為0.8 mL/min,檢測柱溫為30 ℃。
1.3.5 MIPs固相萃取柱的制備及優化
采用干法裝柱的方式,MIPs的填柱量為30 mg,裝入固相萃取柱中,壓實填料后制成分子印跡固相萃取柱(molecularly imprinted solid phase extraction column,MISPE)。以撲草凈為例,考察不同洗脫條件下,MISPE對其吸附能力。采用6 mL乙腈-水溶液(3∶1,V/V)對所制備的萃取柱進行活化;制備4 種三嗪類農藥混合標準溶液,取1 mL進行上樣;以3 mL甲苯溶液進行淋洗;3 mL 5%乙酸-乙腈溶液進行洗脫。獲得的溶液經氮氣吹干后復溶于1 mL乙腈中。采用同樣操作,以常規C18固相萃取柱作為對照。
1.3.6 方法學評價
將含有三嗪類殺蟲劑的黃瓜、蘋果和玉米樣品切碎,分別取5 g,向其中加入20 mL乙腈并充分混勻,重復離心合并上清液。取1 mL氮氣吹干,再溶于等量乙腈-水溶液,依照優化后的MISPE制備方法進行HPLC檢測。
1.3.7 吸附再生性實驗
采用所制備的雙模板MISPE,重復檢測4 種三嗪類殺蟲劑,以50 μg/L乙腈溶液洗脫共20 次,其中每5 次測定其三嗪類農藥的含量。
1.4 統計分析
2 結果與分析
2.1 雙模板分子印跡聚合物制備條件優化
由于不同模板分子間含有固定形狀的孔穴,其獨特的化學性質決定模板分子的特異性吸附功能,所以在預組裝階段中,模板分子之間的配比會使最終的共聚物產生不同的結合位點與吸附活性[22]。表1表明,滅蠅胺和三聚氰胺這兩種模板分子物質的量之比為3∶2時,所制備的印跡聚合物吸附量最大(2.36~2.93 mg/g)。以乙腈作為制孔劑時,其聚合物吸附量(0.71~2.93 mg/g)高于三氯甲烷(0.42~1.84 mg/g),且易研磨,因此選用乙腈作為制孔劑。交聯劑可選用二元交聯劑EGDMA和三元交聯劑TRIM,它們分別與模板分子按物質的量比1∶20與1∶4進行交聯制備印跡聚合物,比較發現選取TRIM為交聯劑時MIPs的吸附量更優,這歸因于其更易產生聚合度更強的交聯網狀結構[23]。因此,最終的制備體系選用滅蠅胺和三聚氰胺3∶2混合為雙模板分子,MAA為功能單體,TRIM為交聯劑,且三者的物質的量比為1∶4∶4,此條件下MIPs識別性能和吸附量最佳。
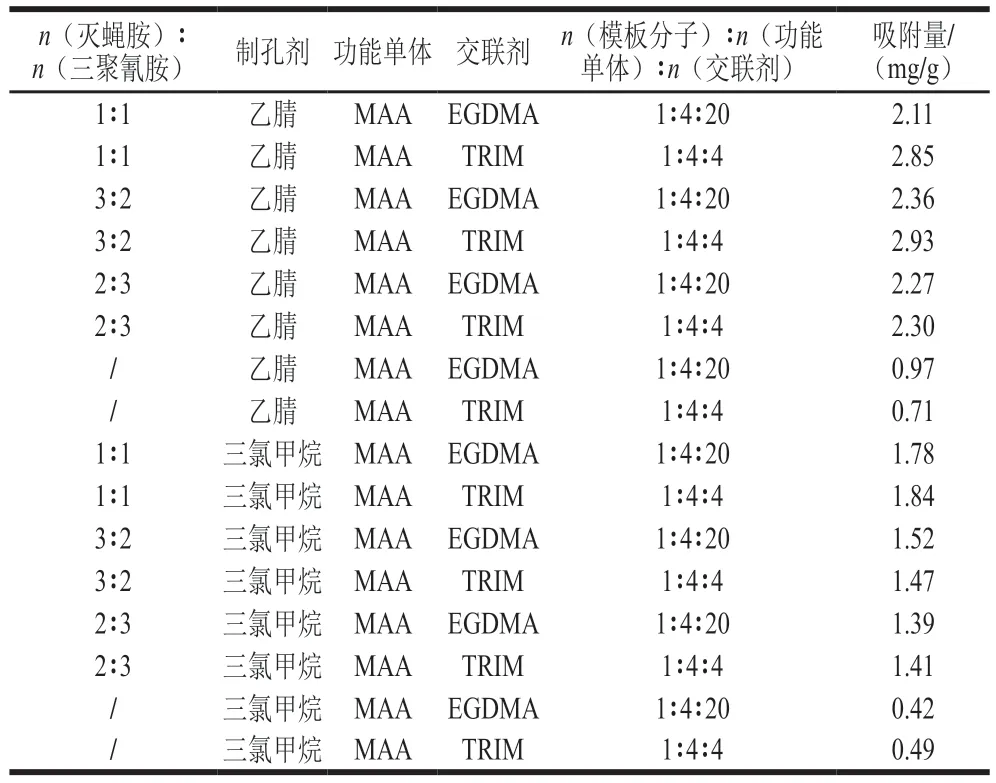
表1 聚合體系的設計和優化Table 1 Design and optimization of polymerization system
2.2 MIPs的形態結構表征
2.2.1 紅外光譜分析
采用紅外吸收光譜實驗對MIPs、NIPs以及模板分子三聚氰胺的表面化學特征進行表征與比較,結果如圖1所示。MIPs與NIPs均在1733、1455、1155 cm-1處分別含有C=O、O—H和C—O—C 3 個特征峰,證明成功進行本體聚合反應。除此之外,對于MIPs來說,在1551 cm-1處已經沒有三聚氰胺的特征吸收峰,說明其中的模板分子已完全洗脫[24]。
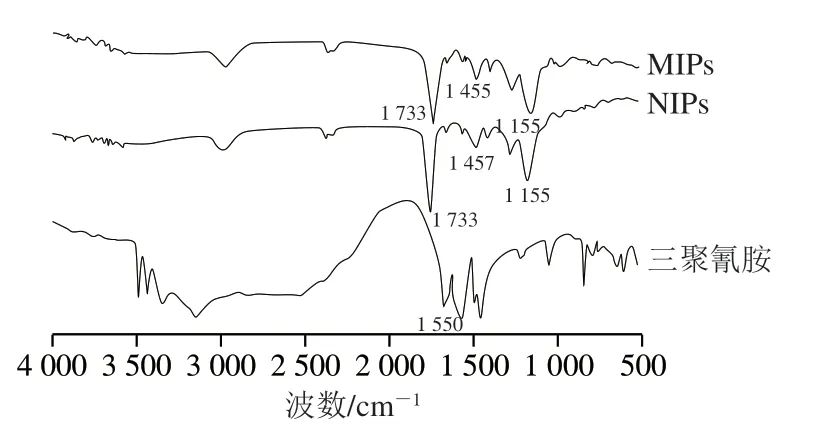
圖1 MIPs、NIPs和三聚氰胺的紅外光譜圖Fig.1 Infrared spectra of MIPs,NIPs and melamine
2.2.2 MIPs和NIPs的掃描電鏡分析
為獲得聚合物的微觀形貌,利用掃描電鏡對MIPs(圖2A)和NIPs(圖2B)進行更進一步觀測。結果顯示,與NIPs相比,MIPs聚合物顯示成微球狀,其表面褶皺程度更為豐富。可認為MIPs的比表面積更大,且結合位點更多,且宏觀表現上其吸附效果優于NIPs。而對于NIPs來說,其中并無模板分子印跡,因此其分子結合形態呈不規律狀。

圖2 MIPs(A)和NIPs(B)的掃描電鏡形態(×30000)Fig.2 Scanning electron micrographs of MIPs (A) and NIPs (B) (× 30000)
2.3 三嗪類MIPs的吸附性能
2.3.1 靜態吸附實驗及Scatchard模型擬合分析
通常靜態吸附實驗可以用來判斷MIPs的吸附能力,如圖3所示。圖3A表明,隨著三嗪農藥撲草凈的濃度增加,MIPs和NIPs對其吸附能力都明顯增強,說明兩者都具有一定水平的物理吸附能力。然而在各個質量濃度下,MIPs比NIPs具有更高的吸附量。可能由于MIPs聚合物分子中產生與三嗪類農藥分子結構特異性結合的孔穴,產生了額外的物理吸附與化學吸附。而對于不含特異性結合位點的NIPs來說,其吸附能力相對較差[25]。
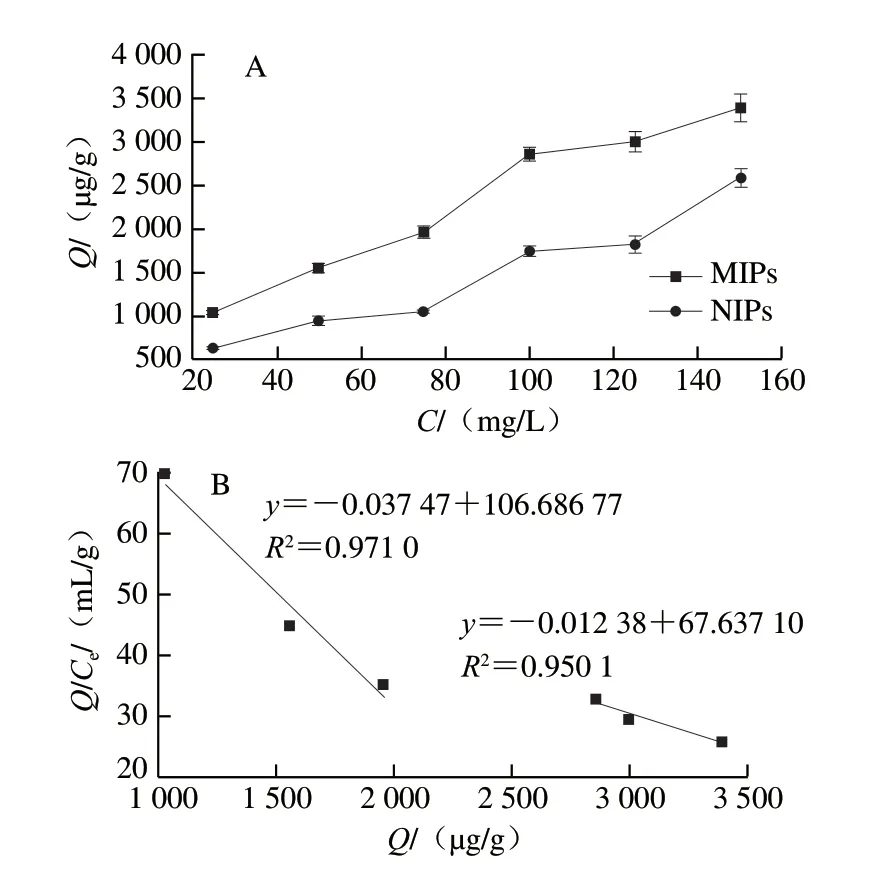
圖3 MIPs和NIPs的靜態吸附實驗(A)及Scatchard模型(B)分析Fig.3 Static adsorption characteristics (A) and Scatchard model analysis (B) of MIPs and NIPs
圖3B為聚合物的Scatchard模型擬合圖,圖中各點并不呈線性,但進行線性擬合后,可看成是斜率不同的兩條線性直線,證實所制備的MIPs中含有兩個特異性結合位點[26]。通過兩個擬合線性方程的斜率和截距,計算出該聚合物的兩種吸附位點的最大吸附量分別為2.85、5.46 mg/g,結合常數分別為26.688、80.775 mg/L。
2.3.2 吸附動力學分析
動態吸附及動力學模型通常用于反映吸附反應的速率及隨時間變化的規律,由圖4可知,在前100 min,吸附曲線斜率比較大,說明MIPs與NIPs吸附比較快速,且隨著時間延長吸附量呈上升趨勢。而在100 min之后,MIPs與NIPs的吸附曲線較為平緩,都逐步達到吸附平衡狀態。然而,在吸附過程中,MIPs表現出比NIPs更快的吸附速度,而且在吸附平衡時其對三嗪類農藥撲草凈的吸附量也更高,這是由MIPs的特定孔穴所導致的特異性吸附[27]。
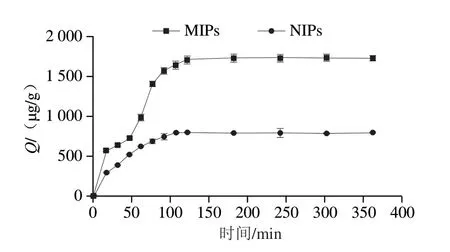
圖4 MIPs和NIPs的吸附動力學Fig.4 Adsorption kinetics of MIPs and NIPs
2.3.3 選擇性吸附實驗分析
對7 種典型三嗪類農藥進行選擇性吸附,MIPs與其對照NIPs對農藥的吸附量(Q)、印跡因子(imprinting factors,IF)和選擇性指數(selectivity index,SI)如表2所示。不同種類農藥的IF均大于1,說明MIP對相應農藥的吸附作用是NIPs的1 倍以上。吡蟲啉屬于氯化煙酰類殺蟲劑,由表2可知,MIPs與NIPs對其吸附量大體一致,證實MIPs對三嗪類殺蟲劑呈現出更優良的選擇性。此外,由于吡蟲啉是含氮結構類似物,和模板分子相比具有不同的結構,無法適應聚合物中的特異性孔穴,故吸附率最低[28]。滅蠅胺和三聚氰胺雙模板MIPs可以增強吸附能力,是由于兩者在和交聯劑發生共聚反應時產生了協同作用,優化了印跡孔穴,從而呈現出最匹配的結合狀態[29]。此外,在模板分子聚合物中形成孔穴時,體積大的模板分子有助于大空隙的形成,從而更利于分子吸附進程[30]。
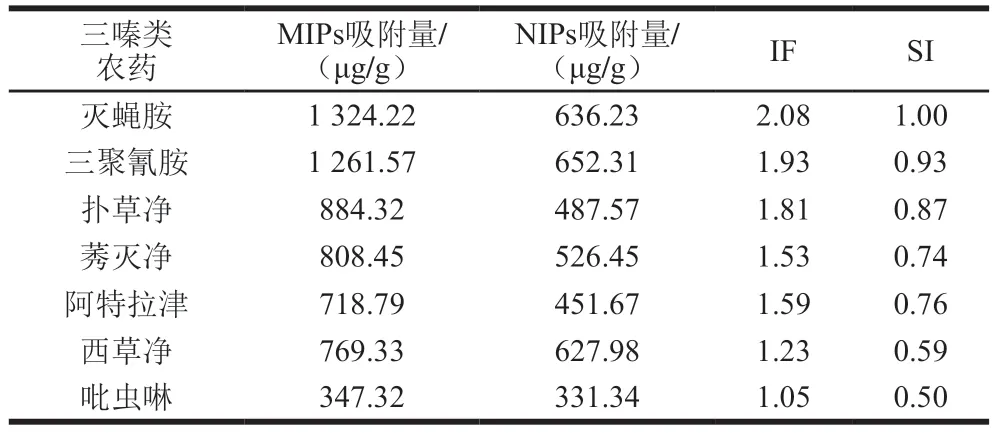
表2 聚合物對不同底物的印跡因子與選擇性指數Table 2 IF and SI of polymers for different pesticide compounds
2.4 雙模板分子印跡固相萃取條件的優化
為優化淋洗條件,選用甲苯、乙腈、二氯甲烷、水為淋洗液,HPLC檢測后得到目標分析物的回收率,如圖5所示。當乙腈和二氯甲烷作為淋洗液時,三嗪類農藥的回收率相對最低。而甲苯作為淋洗劑時,回收率相對最高,淋洗損失量最少。這是由于甲苯中甲基較小,所引起的極性非常弱,而且其溶解性比較好,能夠減少三嗪類農藥的淋洗損失[31]。
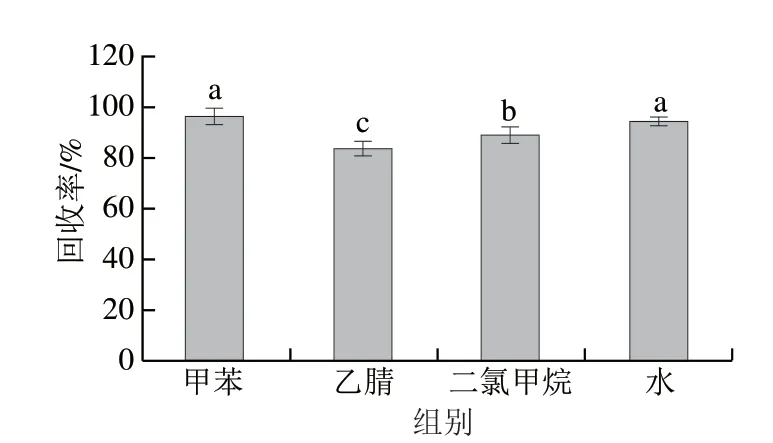
圖5 不同淋洗液對固相萃取回收率的影響Fig.5 Influence of different single eluents on the recovery of solid phase extraction
同時,洗脫液的優化也有助于分析物后續的洗脫過程,一般選用極性較強的溶劑[32]。分別選擇乙腈、甲醇、乙酸-甲醇(5%、10%)、乙酸-乙腈(5%、10%)作為洗脫液,對目標分析物分別洗脫后經HPLC檢測。如圖6所示,選取5%乙酸-乙腈時,回收率最高,說明其洗脫效果最好。
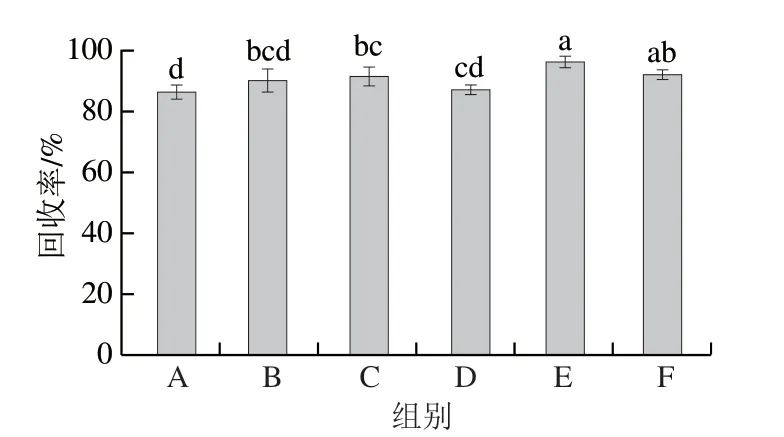
圖6 不同洗脫液對固相萃取回收率的影響Fig.6 Influence of different binary eluents on the recovery of solid phase extraction
通過條件優化,確定最終MIPs的填柱量為30 mg,裝入固相萃取柱后制成MISPE柱。3 mL乙腈和等體積水配比作為活化液,取1 mL上樣液,以3 mL甲苯溶液為淋洗液,3 mL 5%乙酸-乙腈溶液為洗脫液,進行HPLC檢測,以獲得最大的回收率。
2.5 方法學評價
2.5.1 線性范圍與檢出限
配制含4 種三嗪類農藥的混合標準溶液,按照優化的條件進行HPLC測定,所得線性方程的相關參數如表3所示。這些三嗪類農藥線性方程的線性系數均大于0.99,線性范圍均在0.05~1.00 μg/mL之間。阿特拉津的檢出限為0.02 ng/mL,西草凈為0.03 ng/mL,撲草凈和莠滅凈分別為0.01 ng/mL。

表3 4 種三嗪類農藥的線性方程參數與檢出限Table 3 Linear relationships and detection limits of four triazine pesticides
2.5.2 常規固相萃取與分子印跡固相萃取的檢測
以黃瓜加標樣品的(0.1 μg/mL)檢測為例,分別對MISPE柱和C18柱的吸附能力進行檢測,結果見圖7。結果表明,與C18柱相比,經MISPE柱萃取過后,降低了黃瓜中色素等雜質的含量,同時也實現了對三嗪類農藥的富集。
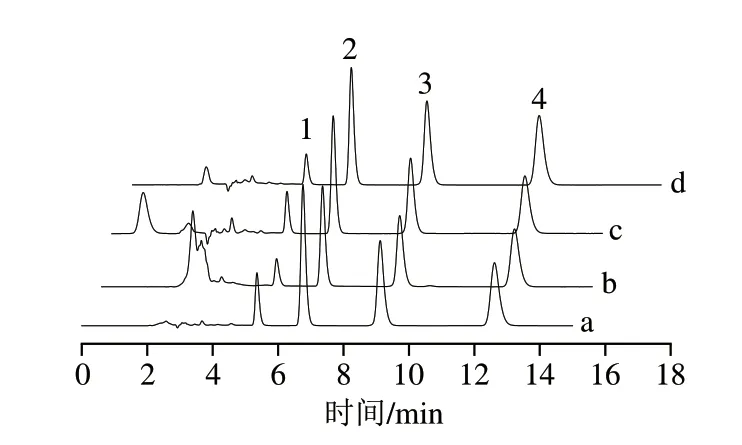
圖7 加標黃瓜樣品提取圖Fig.7 Extracted ion chromatograms of spiked cucumber samples
2.5.3 回收率與精密度實驗
為驗證雙模版MIPs固相萃取-HPLC聯用對三嗪類殺蟲劑的測定效果,分別檢測3 種果蔬樣品中三嗪類農藥,得到平均回收率和相對標準偏差進行測定,結果見表4。在加標質量濃度為0.1、0.25、0.5 μg/mL時,黃瓜樣品中阿拉特津、撲草凈、莠滅凈和西草凈的平均回收率均在85.1%~102.3%之間,相對標準偏差在2.3%~6.8%之間。蘋果樣品中各分析物的平均回收率在75.2%~95.2%,相對標準偏差在2.4%~5.7%之間;玉米樣品中平均回收率在85.2%~95.3%之間,相對標準偏差在2.6%~5.1%之間。由此可知,該雙模版MISPE柱滿足果蔬中三嗪類農藥的特異性檢測要求,檢出限低、精密度高。
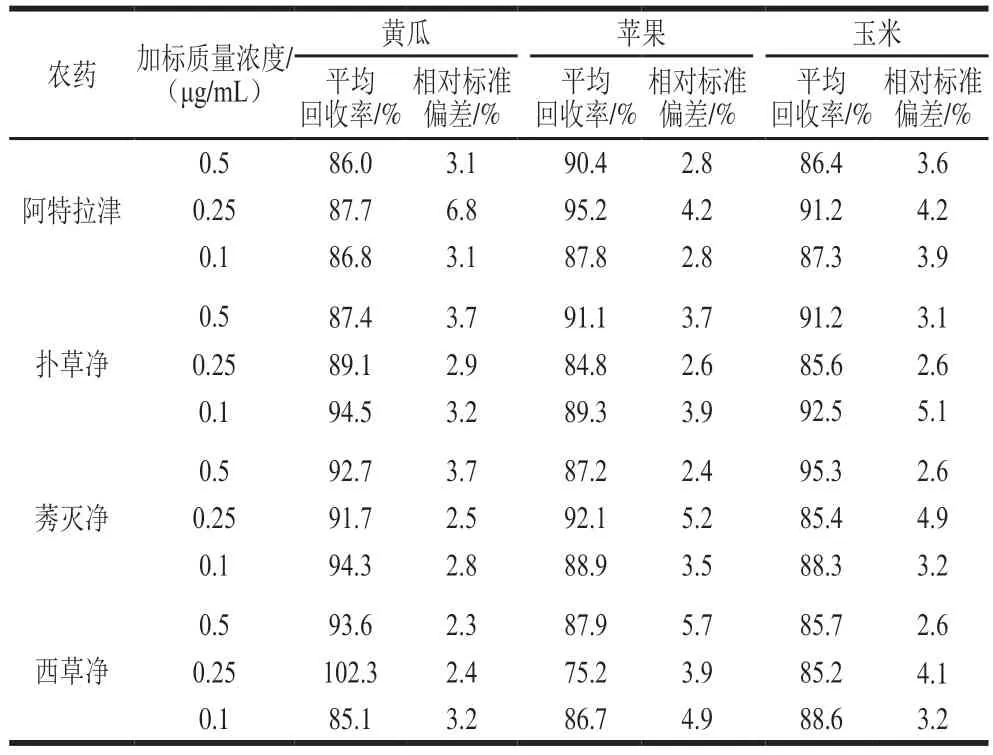
表4 回收率和精密度實驗Table 4 Recoveries and precision RSD
2.5.4 重復使用實驗
采用所制備的MISPE柱進行重復上樣,共20 次,其中每隔5 次檢測洗脫液中的阿拉特津、撲草凈、莠滅凈和西草凈4 種三嗪類農藥含量,回收率結果見表5。重復使用15 次后,回收率仍保持在80%以上,說明MISPE柱可重復性好。

表5 分子印跡固相萃取柱的重復利用率Table 5 Reusability of MISPE%
2.5.5 材料性能評價
將本檢測方法與其他測定三嗪類除草劑的方法進行比較,表6顯示出吸附劑類型、檢測方法、檢出限、回收率等方面的對比。與其他報道的方法相比,該方法制備的吸附材料具有較好的回收率和相似/更低的檢出限,表明該方法具有靈敏度高、精確度好、檢出限低的優點。
3 結論
以滅蠅胺和三聚氰胺為雙模板分子,其物質的量之比為3∶2,且采用MAA為功能單體,TRIM為交聯劑時,制備的雙模版分子印跡聚合物材料MIPs的吸附性能為最優。對其吸附性能進行評價,表現出比NIPs更高的吸附量和良好的選擇性,說明兩種模版分子在和交聯劑相互作用產生共聚反應時,產生協同作用。通過聯用MIPs固相萃取-HPLC法以檢測黃瓜、蘋果和玉米中4 種三嗪類農藥,在加標質量濃度為0.1~0.5 μg/mL時,平均回收率為83.2%~102.3%,標準標準偏差為2.3%~6.8%,且檢出限較低。該方法簡單、準確,明顯增強了檢測效率和精密度,能適用于三嗪類農藥的痕量檢測,在農藥殘留的定性定量分析中具有較好的應用前景。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
