小麥粒重相關性狀的QTL定位及分子標記的開發
2023-11-18 07:09:00張澤源李玥趙文莎顧晶晶張傲琰張海龍宋鵬博吳建輝張傳量宋全昊簡俊濤孫道杰王興榮
中國農業科學 2023年21期
張澤源,李玥,趙文莎,顧晶晶,張傲琰,張海龍,宋鵬博,吳建輝,張傳量,宋全昊,簡俊濤,孫道杰,王興榮
小麥粒重相關性狀的QTL定位及分子標記的開發
張澤源1,李玥2,趙文莎1,顧晶晶3,張傲琰1,張海龍1,宋鵬博1,吳建輝1,張傳量1,宋全昊4,簡俊濤5,孫道杰1,王興榮2
1西北農林科技大學農學院,陜西楊凌 712100;2甘肅省農業科學院作物研究所,蘭州 730000;3洛陽市農林科學院,河南洛陽 471023;4駐馬店市農業科學院,河南駐馬店 463000;5南陽市農業科學院,河南南陽 473000
【目的】小麥是世界總產量第二的糧食作物,而粒重是影響小麥產量的重要因素。以和尚頭(HST)和隴春23(LC23)衍生的216個家系重組自交系(recombinant inbred lines,RIL)群體為材料,基于55K SNP基因型數據,針對小麥粒重相關性狀進行QTL定位,開發和驗證粒長主效QTL的共分離標記,為分子標記輔助選擇育種提供參考。【方法】利用小麥55K SNP芯片對親本和RIL群體進行基因分型,構建高密度遺傳連鎖圖譜,并與中國春參考基因組IWGSC RefSeq v1.0進行相關性分析。基于完備區間作圖法對多環境粒重相關性狀進行QTL定位;通過對主效QTL進行方差分析,判斷不同QTL間的加性互作效應,并分析其對粒重相關性狀的影響。同時,根據粒長主效QTL的共分離SNP位點開發相應的競爭性等位基因特異性PCR標記(kompetitive allele specific PCR,KASP),并在242份國內外小麥種質構成的自然群體中進行驗證。【結果】構建了和尚頭/隴春23 RIL群體的高密度遺傳圖譜,全長4 543 cM,共包含22個連鎖群,覆蓋小麥21條染色體,平均遺傳距離為1.7 cM。遺傳圖譜與物理圖譜具有顯著相關性,Pearson相關系數為0.77—0.99(<0.001)。共檢測到51個粒重相關QTL,其中,有4個為3個及以上環境穩定表達的主效QTL,分布在2D、5A、6B和7D染色體。根據物理區間和功能標記分析主效QTL和分別為光周期基因和開花基因,方差分析表明,二者具有顯著的互作效應;和優異等位基因的聚合顯著提高了小麥的千粒重和粒寬。此外,根據粒長主效位點的共分離SNP開發了相應的KASP分子標記,該標記在242份小麥組成的自然群體中與粒長和粒重性狀顯著相關,在不同環境下能增加粒長3.33%—4.59%(<0.001)和粒重5.70%—10.35%(<0.05)。【結論】和尚頭(HST)和隴春23(LC23)的粒重相關性狀由多個遺傳位點控制,其中,和通過加性互作效應可顯著提高小麥的千粒重和粒寬。與粒重和粒長具有顯著相關性,其共分離分子標記可應用于分子標記輔助選擇育種。
小麥;千粒重;QTL;KASP標記;分子標記輔助選擇育種
0 引言
【研究意義】小麥(L.)是世界35%以上人口的主糧,提供了蛋白質、礦物質和維生素等主要營養元素[1-2]。隨著人口的增加、耕地面積的減少和氣候的變暖,當前的小麥產量已難以滿足人類的需求[3]。因此,發掘小麥產量潛力仍然是育種工作的首要任務。小麥產量的構成要素包括千粒重、每穗粒數和單位面積穗數[4]。其中,千粒重具有較高的遺傳力,可在育種早期世代進行有效的選育[5]。研究表明,千粒重、粒長和粒寬等籽粒性狀與小麥產量呈正相關性[6-7]。因此,明確小麥選育過程中的籽粒相關性狀和基因,對實現高產具有重要的價值和意義。【前人研究進展】盡管栽培小麥的多倍體特性使得數量性狀基因座(QTL)變得復雜,但目前已在小麥21條染色體上發現了大量(400多個)控制粒重和粒型的QTL[8-10]。Ma等[11]以RIL群體基于55K SNP芯片和SSR標記構建了遺傳圖譜,在2D染色體(32.97—33.74 Mb)定位了1個控制粒長、粒寬和千粒重的主效QTL(與連鎖);Qu等[12]利用BSA和小麥660K SNP芯片結合的方法,在2DS染色體上檢測到1個有關粒長和千粒重的共定位區間,物理間距僅為3.97 Mb,并驗證了候選基因在雙親中的差異;Liu等[8]在7D染色體上檢測到與粒重相關的QTL,并定位于3.82 Mb物理區間,其候選基因在第三個外顯子有一個1 bp的插入/缺失(InDel);Yang等[13]通過對2 230個產量相關的QTL進行元分析,發現與粒重相關的QTL分布在小麥的21條染色體上。迄今,已有45個小麥粒重相關基因被報道[14-15],分布在除1D、3B和4B染色體之外的所有染色體上。小麥光周期基因和春化基因也會影響粒重的相關性狀[16]。光周期基因是控制光周期特性的主要基因,可編碼與擬南芥PRR7具有序列相似性的蛋白質,有3個同源基因,分別是、和,主要通過啟動子區域的缺失或插入導致光周期特性的改變[17-18];不同光周期特性與不同緯度氣候條件相適應,可使小麥避開惡劣環境的危害而充分利用光照資源,提高小麥豐產性和穩產性。根據拷貝數不同,將春化基因分為、、和[19]。小麥為成花促進因子,與擬南芥和大麥同源,被命名為[20]。低溫和長日照條件能促進的表達,并通過影響調節開花期,進而顯著影響小麥的產量[20-21]。此外,1B/1R易位系中來自黑麥的1RS染色體不僅含有許多抗病基因,還對小麥的粒重存在顯著影響[22]。【本研究切入點】和尚頭是甘肅干旱地區地方品種,隴春23是甘肅省農業科學院作物研究所和國際玉米小麥改良中心(CIMMYT)創制的小麥品種,二者在粒重和粒型上具有顯著差異(和尚頭的各性狀值均高于隴春23),但其籽粒的遺傳基礎尚不清楚。【擬解決的關鍵問題】本研究以和尚頭/隴春23衍生的重組自交系群體為試驗材料,構建高密度遺傳圖譜,解析和尚頭和隴春23粒重相關性狀的遺傳基礎,發掘粒重和粒型相關QTL位點,并開發相應的高通量KASP檢測標記,為小麥分子輔助選擇育種提供參考。
1 材料與方法
1.1 田間種植與表型鑒定
以和尚頭(HST)和隴春23(LC23)衍生的216個家系F2:8RIL群體和242份國內外小麥品種(系)為試驗材料。HST是甘肅干旱地區的地方品種[23],LC23是由甘肅省農業科學院作物研究所和國際玉米小麥改良中心(CIMMYT)創制的小麥品種[24]。將RIL群體分別種植于陜西楊陵(E1)、甘肅張掖(E2)、河南南陽(E3)和河南洛陽(E4)。隨機區組設計3行區,行長2 m,行距25 cm,株距10 cm,按照常規標準進行田間管理。
小麥成熟后對中間行進行隨機取樣,選擇自然風干的種子,利用萬深SC-G型自動種子考種儀進行粒長、粒寬、籽粒長寬比和千粒重的測量,取單環境平均值和多環境最佳線性無偏預測值(BLUP),用于表型及遺傳分析。
1.2 基因型鑒定
利用Affymetrix? Axiom平臺的小麥55K SNP芯片對216個家系進行全基因組掃描,利用Affymetrix的Axiom Analysis Suite軟件對原始數據進行深入分型。根據Liu等[8]使用的標記合成引物。通過55K SNP芯片獲取的側翼序列,使用PolyMarker在線平臺(https://polymarker.tgac.ac.uk/)設計KASP引物,并在其5′端連接FAM或HEX熒光接頭序列(FAM接頭序列:5′-GAAGGTGACCAAGT TCATGCT-3′;HEX接頭序列:5′-GAAGGTCGGAGTC AACGGATT-3′)。KASP反應體系為2 μL DNA、0.0448 μL引物混合物、2 μL HiGeno 2x Probe Mix B和1.9552 μL ddH2O。反應程序為95 ℃ 10 min;95 ℃ 30 s,65—55 ℃ 25 s,10個循環(每循環降低1.0 ℃);95 ℃ 30 s,55 ℃ 30 s,35個循環;4 ℃避光保存。反應結束后,用酶標儀FLUOstar Omega進行熒光掃描,并用KlusterCaller軟件進行基因分型。SSR標記()的PCR反應體系為DNA 1 μL、2×Rapid Taq Master Mix 10 μL、上下游引物各1 μL和ddH2O 7 μL。PCR反應程序為95 ℃ 5 min;95 ℃ 30 s,53 ℃(1B/1R為56 ℃) 30 s,72 ℃ 30 s,30個循環;72 ℃ 5 min,4 ℃保存。
1.3 數據處理及遺傳圖譜的構建和定位
利用R中的lem4包進行遺傳力計算,公式[25]為。利用Microsoft Excel統計表型數據,利用SPSS 22[26]進行方差分析、檢驗、檢驗和相關性分析。依據株系雜合率(>20%)、株系缺失率(>20%)、基因型缺失率(>20%)和偏分離率(<0.001)等參數篩選55K SNP基因分型數據,利用QTL IciMapping 4.2[27]軟件的BIN功能對剩余SNP標記進行去冗余,獲得Bin標記;利用JoinMap 4.0[28]LOD≥5的Kosambi函數對Bin標記構建連鎖群;根據LOD值結果,使用QTL IciMapping 4.2軟件的MAP功能對SNP標記排序,選用Kosambi函數轉化遺傳距離;最后使用Mapchart 2.3[29]軟件繪制QTL遺傳圖譜。基于完備區間作圖法(ICIM-ADD)的BIP和環境互作QTL(MET)功能進行多環境QTL定位,步長為1.0 cM,臨界值為0.001,使用LOD=3.0作為檢測閾值。
2 結果
2.1 粒重相關性狀的表型鑒定
通過對親本和尚頭和隴春23進行表型鑒定,發現和尚頭的千粒重、粒長、粒寬和籽粒長寬比均高于隴春23(表1),且在RIL群體中出現連續變異和超親分離現象,表明籽粒相關性狀存在多基因遺傳,以及在雙親中均存在優異的QTL等位基因。其中,千粒重和籽粒長寬比的遺傳力較高,分別為0.81和0.84。
通過對粒重相關性狀進行分析(圖1),粒寬與千粒重、粒長呈極顯著相關性(=0.65和0.67,<0.001);粒長與千粒重、籽粒長寬比呈顯著相關性(= 0.48和0.32,<0.001);而籽粒長寬比與千粒重、粒寬呈負相關性(=-0.29和-0.49,<0.001)。在不同環境間,千粒重和籽粒長寬比呈極顯著相關性(= 0.45—0.66,<0.001);在南陽和洛陽試驗點,粒長和粒寬相關性不顯著,但在其他環境中均呈極顯著相關性(=0.26—0.45,<0.001)(附圖1)。表明在群體中可能存在粒重和粒型性狀的主效遺傳位點。
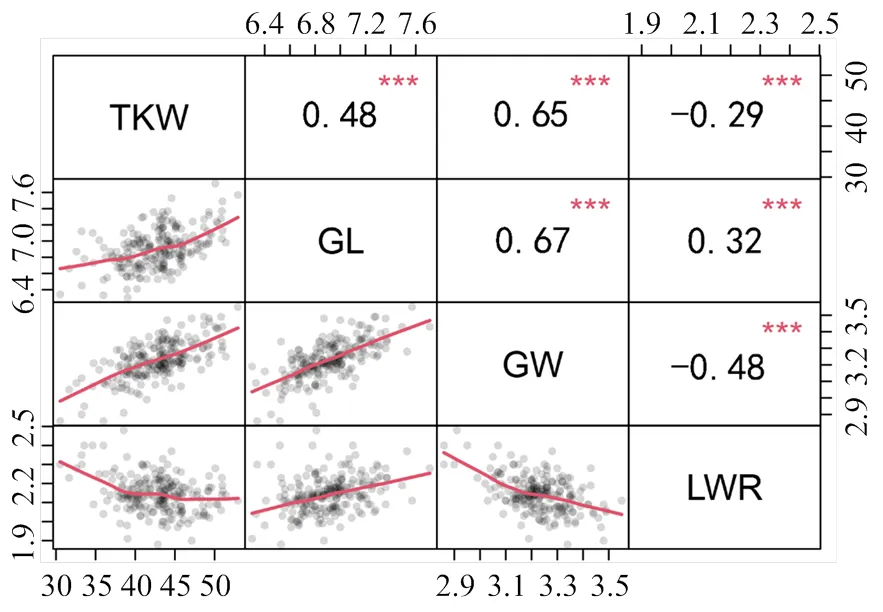
***:P<0.001。下同 The same as below
E1:陜西楊凌;E2:甘肅張掖;E3:河南南陽;E4:河南洛陽;TKW:千粒重;GL:粒長;GW:粒寬;LWR:籽粒長寬比;BLUP表示最佳線性無偏預測值;*和**分別表示在<0.05和<0.01水平差異顯著。下同
E1: Yangling, Shaanxi; E2: Zhangye, Gansu; E3: Nanyang, Henan; E4: Luoyang, Henan;TKW: 1000-grain weight; GL: grain length; GW: grain width; LWR: grain length-width ratio; BLUP represents best linear unbiased prediction; * and ** indicated significant difference at<0.05 and<0.01. The same as below
2.2 遺傳圖譜的構建及共線性分析
通過對原始數據過濾,共獲得16 529個SNP標記,利用BIN功能去冗余后,獲得2 672個Bin標記,構建遺傳圖譜,其全長4 543 cM,包含22個連鎖群,Bin標記之間的平均遺傳距離為1.70 cM,最大遺傳距離為31.82 cM(6D染色體),覆蓋小麥21條染色體(表2),每條染色體上的Bin標記數目不等。7A染色體由2個連鎖群組成,其余染色體均為一個連鎖群。此外,5D染色體的遺傳長度最長,為305.92 cM;4B染色體最短,為122.92 cM。位于A、B和D基因組上的Bin標記數分別為1 025、1 023和624個;SNP標記數分別為6 374、6 884和3 271個;遺傳長度分別為1 509.26、1 423.17和1 610.57 cM。
根據參考基因組對遺傳圖譜和物理圖譜進行共線性分析,結果表明,該遺傳圖譜與中國春參考基因組物理圖譜之間具有良好的共線性,標記順序與小麥基因組組裝的標記順序相對一致,相關系數為0.77—0.99(<0.001,圖2)。每條染色體的遺傳重組表現不平衡現象,染色體端粒區域重組率較高,而中部區域重組率較低,整體呈U型分布;染色體的遺傳位置隨著物理位置的增加而增加,兩端斜率較大,使得共線圖整體呈現出S型;每條染色體的Bin標記數目基本都符合兩端較多,而中間較少的特點。整體來看,染色體兩端為重組熱點區,而中間部分為重組冷點區。其中,在4B和5A的中間部分沒有SNP標記的存在(>200 Mb),但依然為同一條連鎖群,說明該區域為重組冷點區。
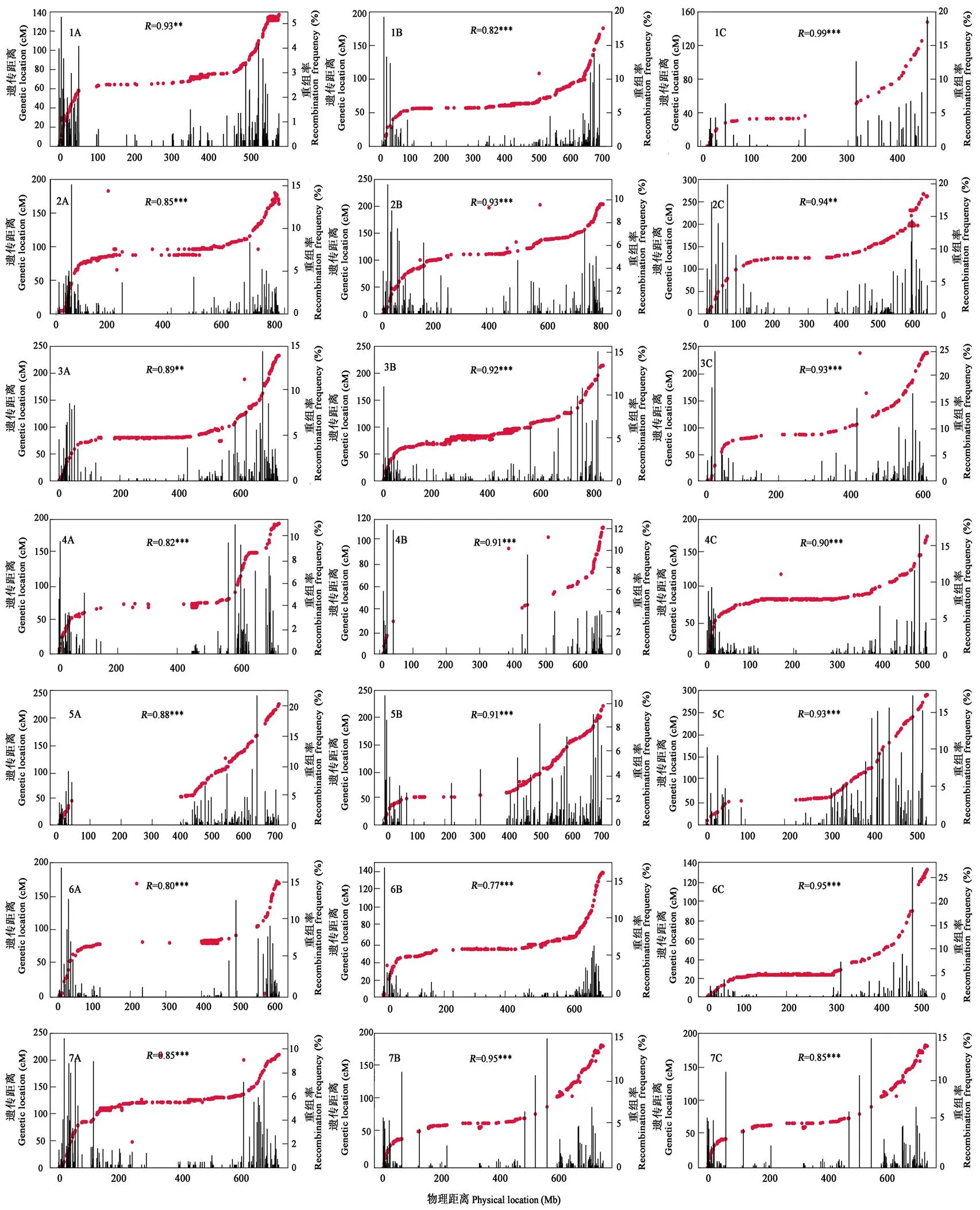
紅色散點表示共線性,黑色直方圖表示Bin標記在參考基因組上的重組率。**:P<0.01,***:P<0.001
2.3 粒重相關性狀的QTL定位
利用和尚頭/隴春23的RIL群體共檢測到51個粒重相關的QTL,單位點可解釋0.44%—20.13%表型變異,LOD值為3.00—59.43。3個及以上環境穩定表達的主效QTL有4個,分布在2D、5A、6B和7D染色體上(表3、圖3和附表1)。
共檢測到9個千粒重QTL,單位點可解釋3.84%— 13.26%表型變異,LOD值為3.16—11.89;其中,(加性效應來自隴春23)和(加性效應來自和尚頭)可在3個以上環境中被檢測到,表型變異解釋率分別為7.19%— 12.92%和7.53%—13.26%,LOD值分別為4.33—11.89和4.63—10.71。
共檢測到13個粒長QTL,單位點可解釋0.44%— 17.86%表型變異,LOD值為3.03—59.43;其中,(加性效應來自和尚頭)可在3個環境中被檢測到,表型變異解釋率為4.56%—17.86%,LOD值為3.93—59.43。
共檢測到15個粒寬QTL,單位點可解釋3.41%— 12.36%的表型變異,LOD值為3.00—16.00;其中(加性效應來自隴春23)和(加性效應來自和尚頭)可在2個環境中被檢測到,表型變異解釋率分別為7.28%— 12.36%和7.85%—20.13%,LOD值分別為4.41—10.14和4.76—16.00。
共檢測到14個籽粒長寬比QTL,單位點可解釋2.37%—14.51%的表型變異,LOD值為3.08—12.93;其中,(加性效應來自隴春23)可在3個環境中被檢測到,表型變異解釋率為4.87%— 11.83%,LOD值為4.87—9.13。
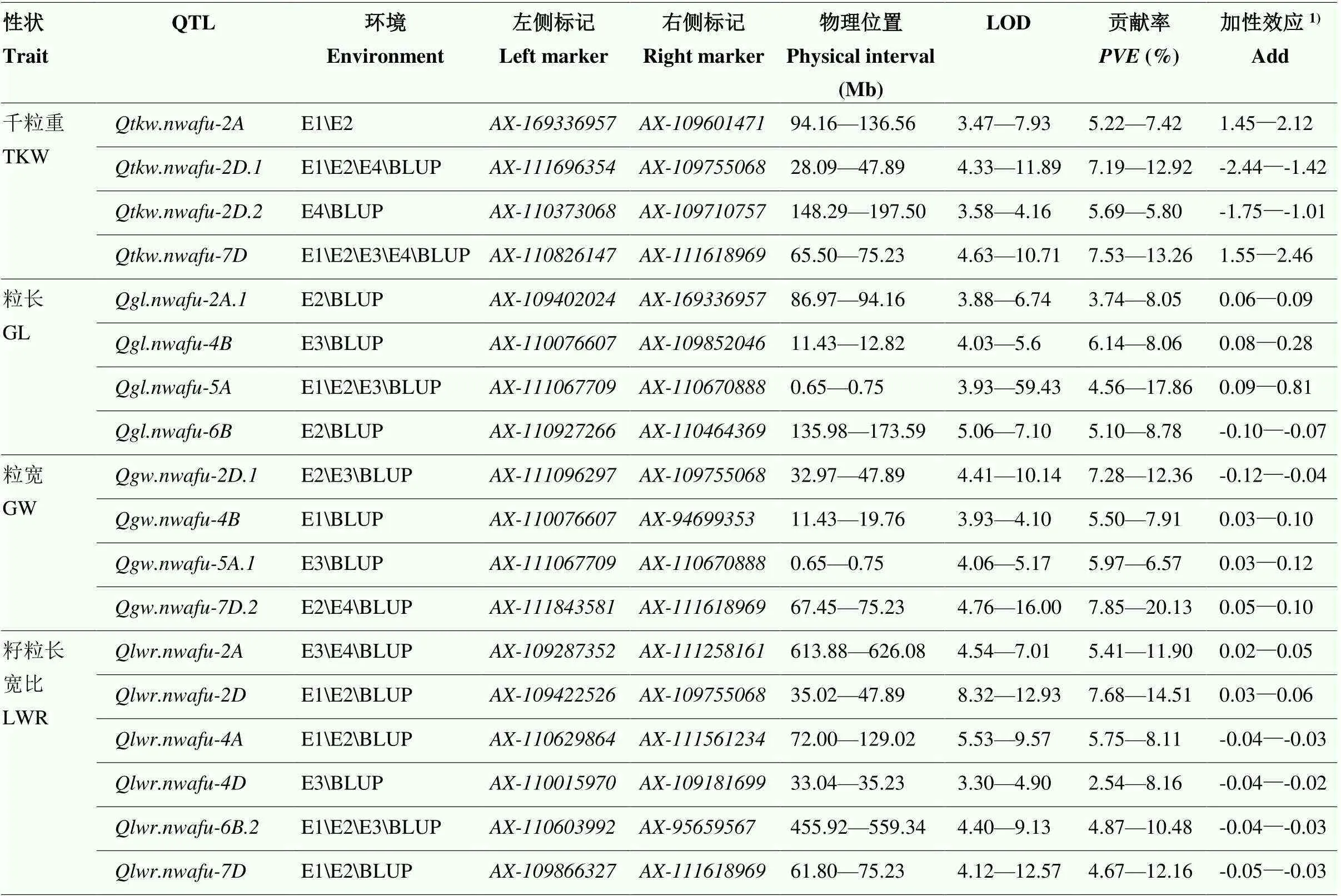
表3 粒重相關性狀的部分QTL
1)加性效應為正說明增效效應來源于和尚頭,加性效應為負說明增效效應來源于隴春23
1)Positive additive effect indicated that the positive allele derived from HST, and negative additive effect indicated that the positive allele derived from LC23
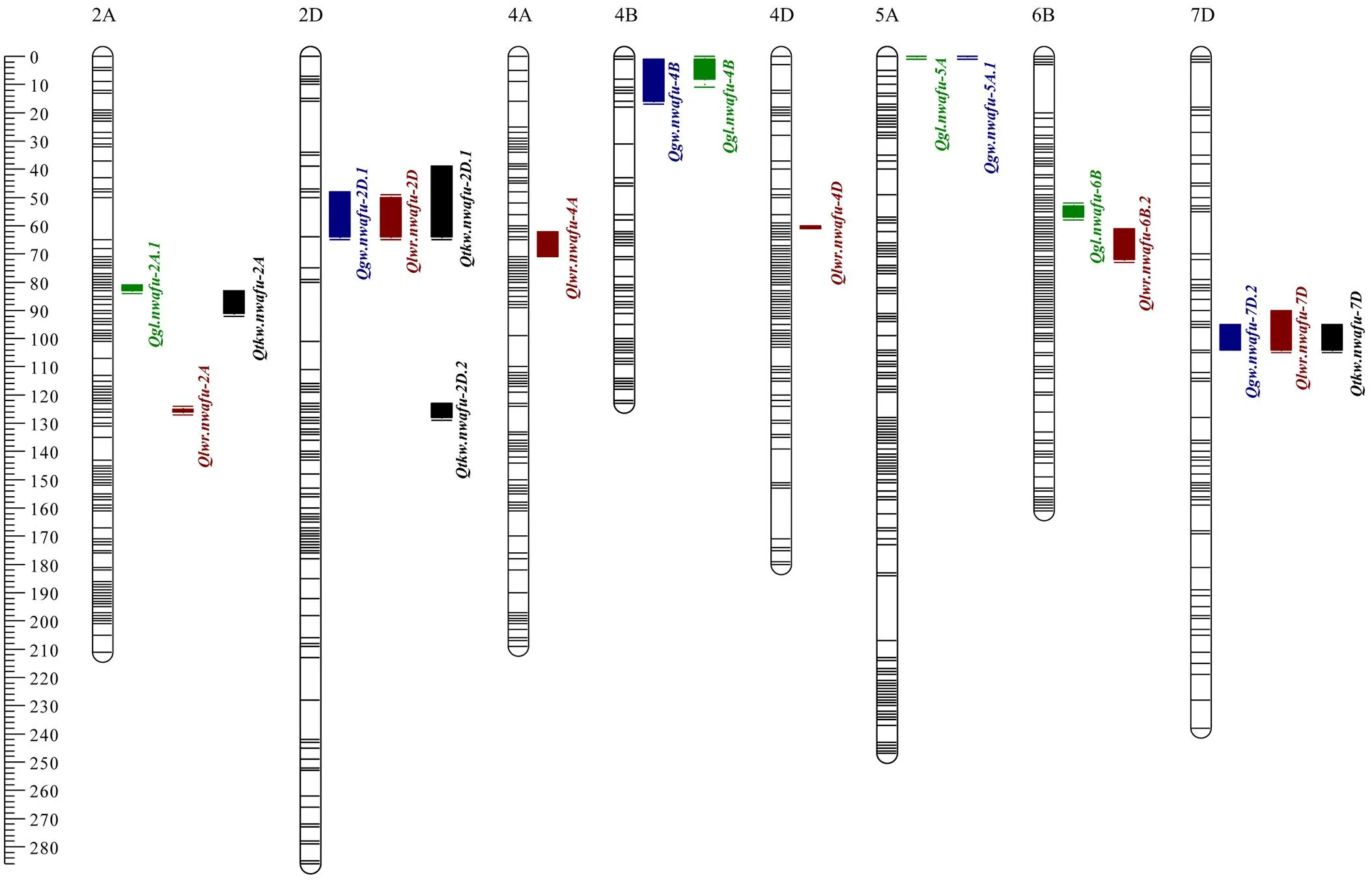
圖3 粒重相關性狀QTL的染色體分布
共發現4個QTL簇,分別位于2D(、和)、4B(和)、5A(和)和7D(、和)染色體上,表明可能存在一因多效QTL。
2.4 環境互作QTL的定位
QTL×環境(QE)互作分析顯示,所有多環境穩定的QTL均能被檢測到,進一步表明QTL的穩定性(附表2)。在QE互作分析中,的總表型變異解釋率為5.76%,其中,加性效應的表型變異解釋率為4.91%,LOD值為19.93;的總表型變異解釋率為6.56%,其中,加性效應的表型變異解釋率為5.14%,LOD值為20.01;的總表型變異解釋率為11.82%,其中,加性效應的表型變異解釋率為6.14%,LOD值為8.88。
2.5 Qtkw.nwafu-2D.1和Qtkw.nwafu-7D的效應分析
在不同環境條件下,攜帶優異等位基因的株系可以提高千粒重6.10%—10.77%(<0.01),增加粒寬3.23%—6.01%(<0.001);攜帶優異等位基因的株系可以提高千粒重4.31%—8.25%(<0.05),增加粒寬4.41%—4.84%(<0.05);然而,同時攜帶和優異等位基因的株系卻對粒長未產生顯著影響。通過進一步探究和對株高和抽穗期的影響,結果表明,在不同環境條件下,攜帶優異等位基因的株系可以降低株高6.32%—6.33%(<0.05),縮短抽穗期3.61%—5.10%(<0.001);攜帶優異等位基因的株系可以降低株高5.39%—6.30%(<0.001),縮短抽穗期1.56%—1.64%(<0.05)(附表3)。
方差分析表明(表4),和存在極顯著的互作效應(<0.01);受環境影響較大,其環境互作對粒寬和粒長有顯著影響(<0.01);和對千粒重、粒寬和籽粒長寬比有顯著影響,對粒長無顯著影響(<0.001)。聚合效應表明(圖4),同時攜帶和優異等位基因株系的千粒重和粒寬可顯著增加13.07%(<0.05)和4.46%(<0.05)。
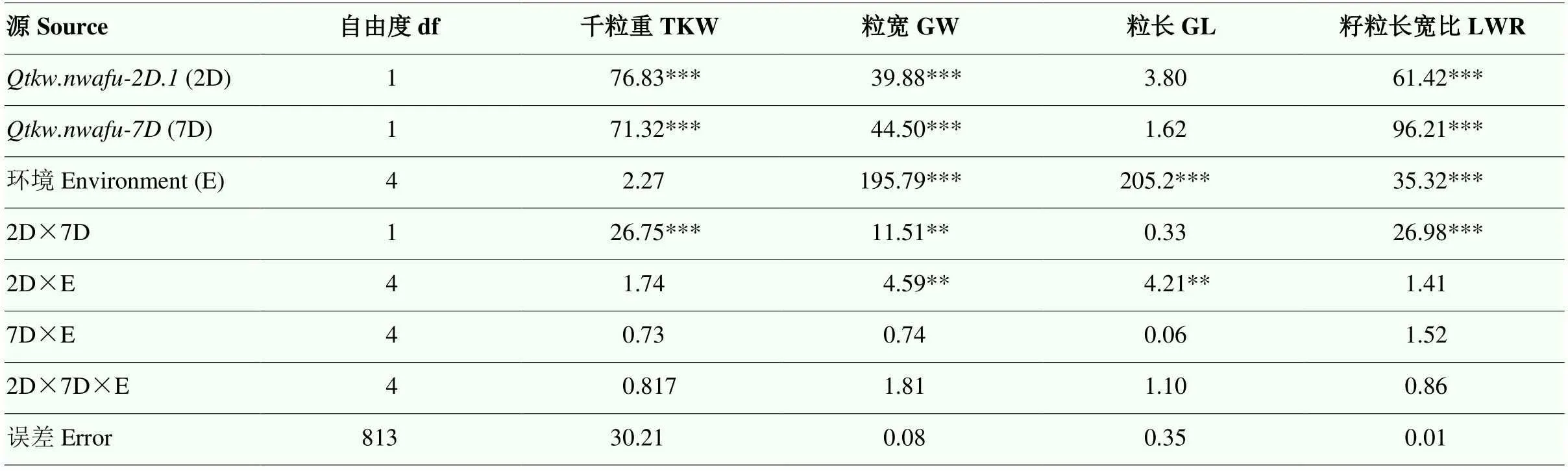
表4 不同環境下Qtkw.nwafu-2D.1和Qtkw.nwafu-7D的方差分析
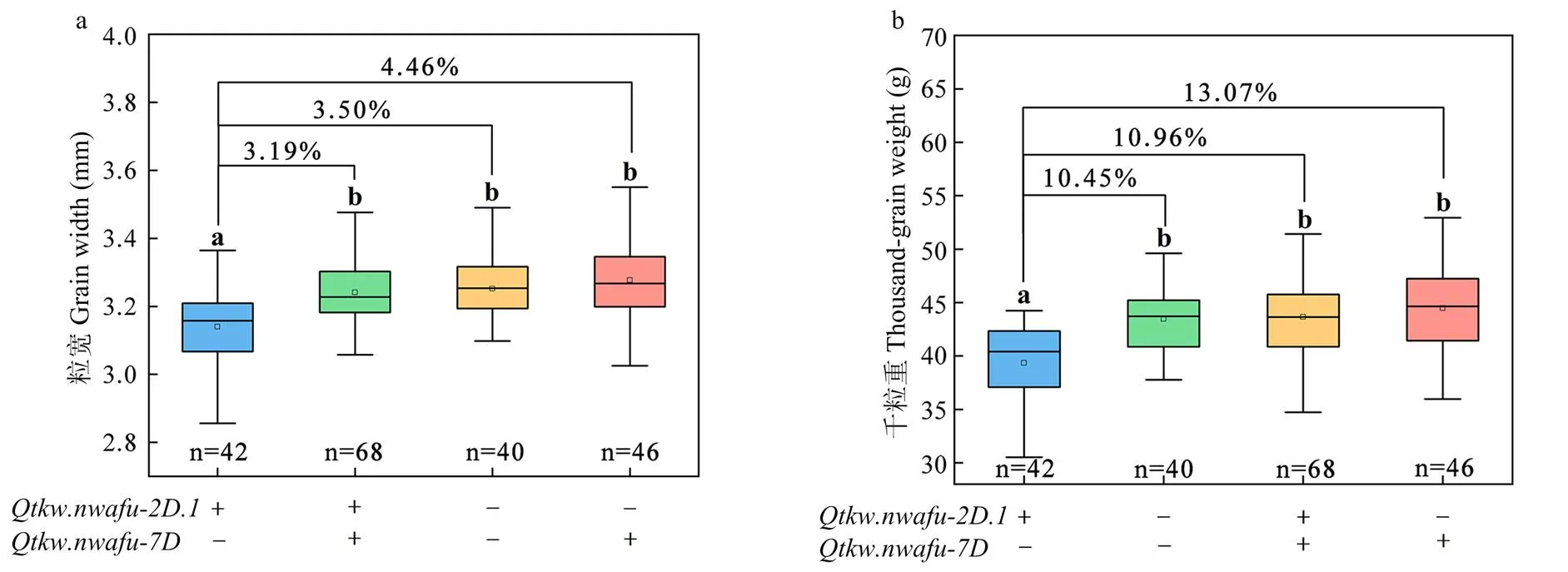
+:相應側翼標記的等位基因來自和尚頭的株系;-:表明相應側翼標記的等位基因來自隴春23的株系。不同小寫字母表示差異顯著
2.6 Qgl.nwafu-5A KASP標記的開發與驗證
根據目標區間兩側的序列開發KASP標記,其中,在RIL群體親本之間和子代之間均具有多態性,利用242份國內外小麥品種(系)驗證位點(附圖2),結果表明,該位點分型明顯,在不同環境條件下可以增加粒長3.33%—4.59%(<0.001)(圖5),增加千粒重5.70%—10.35%(<0.05)(附表3),可用于分子標記輔助選擇育種。
3 討論
3.1 與已知相關基因/QTL位點的比較
六倍體小麥是由野生二粒小麥和節節麥自然雜交形成的,雖然D亞基因組的遺傳變異相對較少,但對六倍體小麥的籽粒大小和形狀具有明顯的影響,尤其是2D和7D染色體對小麥改良起到了積極的正向調節作用[30]。本研究在2D和7D染色體上各檢測到一個QTL簇,包含千粒重、粒寬和籽粒長寬比QTL,說明2D和7D染色體對小麥粒重和粒型具有重要影響。位于SNP標記和之間,根據其物理位置推測,與Ma等[11]、Yu等[31]和Kumar等[6]定位結果一致;位于SNP標記和之間,其物理區間與前人定位結果重合[6, 8, 32-35],且為相同位點。由于和的物理區間分別與已克隆的光周期基因和開花基因重合,故使用和的標記對RIL群體進行檢測,根據分型結果(附圖3和附表4),將標記定位到和之間,因此,推測這兩個位點效應可能與和相關。光周期不敏感型等位基因影響下游基因和的表達,進而促進小麥提前開花和加快抽穗[17];對調節小麥開花起主要作用[20],影響抽穗和籽粒發育[36];與小麥春化基因緊密連鎖[37],且受光周期途徑調控[38],進而相互協調共同參與小麥生長發育。綜上,本研究在和尚頭/隴春23群體中發現的和位點分別與和相關。
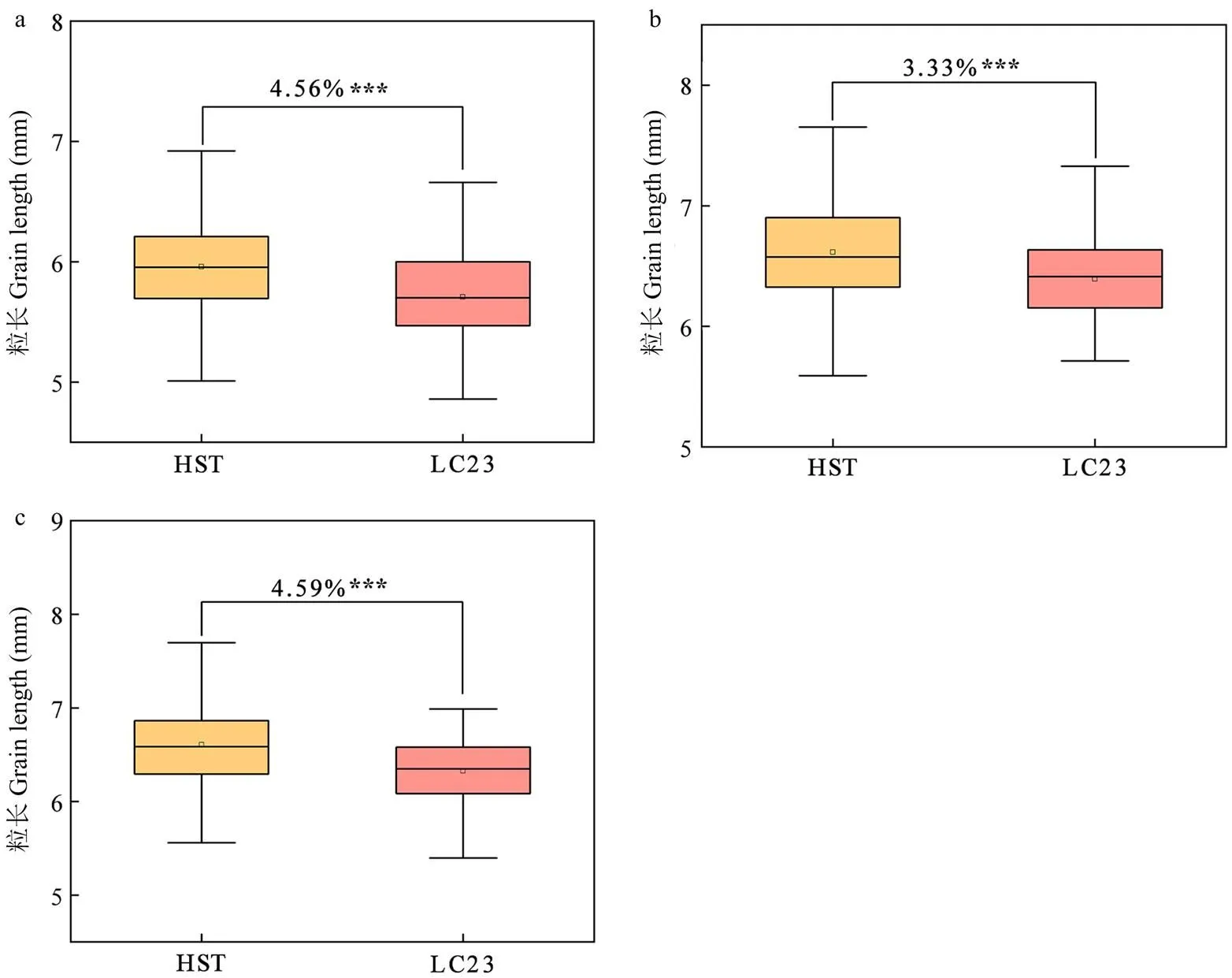
a:楊陵;b:南陽;c:洛陽a: Yangling; b: Nanyang; c: Luoyang
粒長相關位點被定位在分子標記和之間,物理位置為0.65—0.75 Mb,可在3個環境中被檢測到,平均表型變異解釋率為9.76%,是主效QTL。根據前人研究結果,與Wei等[39]在單環境中檢測到的位點部分重合,可能是同一位點。Yu等[31]在2A染色體檢測到的千粒重QTL與區間重疊,該位點可在2個環境中被檢測到,表型變異解釋率為4.3%—13.6%,LOD值為3.5—14.0。其余QTL未被報道,單位點可解釋5.69%—11.90%的表型變異,LOD值為3.30—9.57,均為新QTL。考慮到1B/1R易位系對小麥籽粒性狀的影響,本研究使用1B/1R的SSR功能標記對RIL群體進行檢測,發現雙親及衍生群體中均存在基因型差異,但檢驗結果顯示,1B/1R易位系未對千粒重產生顯著影響。
3.2 主效QTL Qtkw.nwafu-2D.1和Qtkw.nwafu-7D間的互作分析和Qgl.nwafu-5A分子標記的開發
和存在極顯著的QTL互作,可能與共同調控光周期途徑和春化途徑的基因有關[40]。和對千粒重、粒寬、株高和抽穗期均有顯著影響;優良等位基因的聚合是育種家創新種質資源和提高小麥產量的有效途徑[41],在RIL群體中,聚合和位點對千粒重和粒寬均有顯著的加性效應,說明它們可能共同參與小麥生長發育的調節,并對小麥籽粒的發育影響很大,因此,在小麥育種選擇和品種改良中具有重要的應用價值。此外,本研究還發現,與相比,受環境影響更小,這與攜帶品種的千粒重穩定性更好[16]相一致。本研究還開發了的共分離KASP分子檢測標記,并在242份小麥種質中進行了驗證,即該標記與粒重和粒長性狀顯著相關,為的分子輔助選擇育種奠定了基礎。
4 結論
利用和尚頭/隴春23衍生的RIL群體共檢測到51個粒重相關性狀的QTL,其中,2D和7D染色體上存在效應明顯的QTL簇,與千粒重、粒寬、株高和抽穗期等多個性狀顯著關聯。()和()之間存在顯著的加性互作效應,2個位點的聚合能夠促進千粒重和粒寬的提高。針對主效粒長位點開發了共分離的KASP標記,可用于分子標記輔助選擇育種。
致謝:文章得到劉勝杰同學和黃碩同學的幫助,在此表示感謝。
[1] SHEWRY P R, HEY S J. The contribution of wheat to human diet and health. Food and energy security, 2015, 4(3): 178-202.
[2] LI S D, WANG L, MENG Y N, HAO Y F, XU H X, HAO M, LAN S Q, ZHANG Y J, LV L J, ZHANG K, PENG X H, LAN C X, LI X P, ZHANG Y L. Dissection of genetic basis underpinning kernel weight-related traits in common wheat. Plants (Basel, Switzerland), 2021, 10(4): 713.
[3] SHIFERAW B, SMALE M, BRAUN H J, DUVEILLER E, REYNOLDS M, MURICHO G. Crops that feed the world 10. Past successes and future challenges to the role played by wheat in global food security. Food Security, 2013, 5(3): 291-317.
[4] HU J M, WANG X Q, ZHANG G X, JIANG P, CHEN W Y, HAO Y C, MA X, XU S S, JIA J Z, KONG L R, WANG H W. QTL mapping for yield-related traits in wheat based on four RIL populations. Theoretical and Applied Genetics, 2020, 133(3): 917-933.
[5] ZHANG J J, SHE M Y, YANG R C, JIANG Y J, QIN Y B, ZHAI S N, BALOTF S, ZHAO Y, ANWAR M, ALHABBAR Z, JUHáSZ A, CHEN J S, LIU H, LIU Q E, ZHENG T, YANG F, RONG J K, CHEN K F, LU M Q, ISLAM S, MA W Y. Yield-related QTL clusters and the potential candidate genes in two wheat DH populations. International Journal of Molecular Sciences, 2021, 22(21): 11934.
[6] KUMAR A, MANTOVANI E E, SEETAN R, SOLTANI A, ECHEVERRY-SOLARTE M, JAIN S, SIMSEK S, DOEHLERT D, ALAMRI M S, ELIAS E M, KIANIAN S F, MERGOUM M. Dissection of genetic factors underlying wheat kernel shape and size in an Elite×Nonadapted cross using a high density SNP linkage map. Plant Genome, 2016, 9(1): 55.
[7] CAO J J, SHANG Y Y, XU D M, XU K L, CHENG X R, PAN X, LIU X, LIU M L, GAO C, YAN S N, YAO H, GAO W, LU J, ZHANG H P, CHANG C, XIA X C, XIAO S H, MA C X. Identification and validation of new stable QTLs for grain weight and size by multiple mapping models in common wheat. Frontiers in Genetics, 2020, 11: 584859.
[8] LIU H, ZHANG X T, XU Y F, MA F F, ZHANG J P, CAO Y W, LI L H, AN D G. Identification and validation of quantitative trait loci for kernel traits in common wheat (L.). BMC Plant Biology, 2020, 20(1): 529.
[9] PANG Y L, LIU C X, WANG D F, ST AMAND P, BERNARDO A, LI W H, HE F, LI L Z, WANG L M, YUAN X F, DONG L, SU Y, ZHANG H R, ZHAO M, LIANG Y L, JIA H Z, SHEN X T, LU Y, JIANG H M, WU Y Y, LIU S B. High-resolution genome-wide association study identifies genomic regions and candidate genes for important agronomic traits in wheat. Molecular Plant, 2020, 13(9): 1311-1327.
[10] XIN F, ZHU T, WEI S, HAN Y, ZHAO Y, ZHANG D, MA L, DING Q. QTL mapping of kernel traits and validation of a major QTL for kernel length-width ratio using SNP and bulked segregant analysis in wheat. Scientific Reports, 2020, 10(1): 25.
[11] MA J, ZHANG H, LI S Q, ZOU Y Y, LI T, LIU J J, DING P Y, MU Y, TANG H P, DENG M, LIU Y X, JIANG Q T, CHEN G Y, KANG H Y, LI W, PU Z E, WEI Y M, ZHENG Y L, LAN X J. Identification of quantitative trait loci for kernel traits in a wheat cultivar Chuannong16. BMC Genetics, 2019, 20(1): 77.
[12] QU X R, LI C, LIU H, LIU J J, LUO W, XU Q, TANG H P, MU Y, DENG M, PU Z E, MA J, JIANG Q T, CHEN G Y, QI P F, JIANG Y F, WEI Y M, ZHENG Y L, LAN X J, MA J. Quick mapping and characterization of a co-located kernel length and thousand-kernel weight-related QTL in wheat. Theoretical and Applied Genetics, 2022, 135(8): 2849-2860.
[13] YANG Y, AMO A, WEI D, CHAI Y M, ZHENG J, QIAO P F, CUI C G, LU S, CHEN L, HU Y G. Large-scale integration of meta-QTL and genome-wide association study discovers the genomic regions and candidate genes for yield and yield-related traits in bread wheat. Theoretical and Applied Genetics, 2021, 134(9): 3083-3109.
[14] 張香宇. 小麥RHL32籽粒發育相關基因克隆及其與TaRPP13L1多效性功能初析[D]. 楊凌: 西北農林科技大學, 2022.
ZHANG X Y.Cloning of gene related to grain development in wheat RHL32 and pleiotropic function dissection of the candiated genes and TaRPP13L1[D]. Yangling: Northwestern Agriculture and Foresty University, 2022. (in Chinese)
[15] YANG F P, LIU G Y, WU Z Y, ZHANG D X, ZHANG Y F, YOU M S, LI B Y, ZHANG X H, LIANG R Q. Cloning and functional analysis of TaWRI1Ls, the key genes for grain fatty acid synthesis in bread wheat. International Journal of Molecular Sciences, 2022, 23(10): 5293.
[16] 朱雪成. 春化和光周期基因在江蘇小麥品種中的分布及其對農藝性狀的效應分析[D]. 揚州: 揚州大學, 2019.
ZHU X C. Distribution of vernalization and photoperiod genes in Jiangsu wheat cultivars and their effects on agronomic traits [D]. Yangzhou: Yangzhou University, 2019. (in Chinese)
[17] BEALES J, TURNER A, GRIFFITHS S, SNAPE J W, LAURIE D A. Ais misexpressed in the photoperiod insensitive Ppd-D1a mutant of wheat (L.). Theoretical and Applied Genetics, 2007, 115(5): 721-733.
[18] NISHIDA H, YOSHIDA T, KAWAKAMI K, FUJITA M, LONG B, AKASHI Y, LAURIE D A, KATO K. Structural variation in the 5′ upstream region of photoperiod-insensitive alleles Ppd-A1a and Ppd-B1a identified in hexaploid wheat (L.), and their effect on heading time. Molecular Breeding, 2013, 31(1): 27-37.
[19] PUGSLEY A T. A genetic analysis of the spring-winter habit of growth in wheat. Australian Journal of Agricultural Research, 1971, 22(1): 21.
[20] YAN L, FU D, LI C, BLECHL A, TRANQUILLI G, BONAFEDE M, SANCHEZ A, VALARIK M, YASUDA S, DUBCOVSKY J. The wheat and barley vernalization gene VRN3 is an orthologue of FT. Proceedings of the National Academy of Sciences of the United States of America, 2006, 103(51): 19581-19586.
[21] SHIMADA S, OGAWA T, KITAGAWA S, SUZUKI T, IKARI C, SHITSUKAWA N, ABE T, KAWAHIGASHI H, KIKUCHI R, HANDA H, MURAI K. A genetic network of flowering-time genes in wheat leaves, in which an APETALA1/FRUITFULL-like gene, VRN1, is upstream of FLOWERING LOCUS T. The Plant Journal, 2009, 58(4): 668-681.
[22] MORENO-SEVILLA B, BAENZIGER P S, PETERSON C J, GRAYBOSCH R A, MCVEY D V. The 1BL/1RS translocation: Agronomic performance of F3-derived lines from a winter wheat cross. Crop Science, 1995, 35(4): 1051-1055.
[23] 王興榮, 張彥軍, 茍作旺, 李玥, 陳偉英, 祁旭升. 甘肅“和尚頭”小麥調查報告. 甘肅農業科技, 2015(12): 49-52.
WANG X R, ZHANG Y J, GOU Z W, LI Y, CHEN W Y, QI X S. Investigation report of wheat Gansu “Heshangtou”. Gansu Agricultural Science and Technology, 2015(12): 49-52. (in Chinese)
[24] 袁俊秀, 楊文雄. 豐產廣適優質春小麥新品種——隴春23號. 麥類作物學報, 2009, 29(4): 740.
YUAN J X, YANG W X. Longchun 23, a new spring wheat variety with high yield, wide adaptability and high quality. Journal of Triticeae Crops, 2009, 29(4): 740. (in Chinese)
[25] 王建康, 蓋鈞鎰. 利用雜種F2世代鑒定數量性狀主基因-多基因混合遺傳模型并估計其遺傳效應. 遺傳學報, 1997, 24(5): 432-440.
WANG J K, GAI J Y. Identification of major gene and polygene mixed inheritance model and estimation of genetic parameters of a quantitative trait from F2progeny. Acta Genetica Sinica, 1997, 24(5): 432-440. (in Chinese)
[26] ALLEN P, BENNETT K. SPSS statistics version 22: A practical guide. 2014.
[27] MENG L, LI H H, ZHANG Y L, WANG J K. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. The Crop Journal, 2015, 3(3): 269-283.
[28] OOIJEN J, van't Verlaat J W, Tol J, Dale?n J, Buren J, Meer J M, Krieken J V, Kessel J, Van O, Voorrips R, Heuvel L. JoinMap?4, Software for the calculation of genetic linkage maps in experimental populations. Biology, 2006.
[29] VOORRIPS R E. MapChart: Software for the graphical presentation of linkage maps and QTLs. Journal of Heredity, 2002, 93(1): 77-78.
[30] ZHAO G Y, ZOU C, LI K, WANG K, LI T B, GAO L F, ZHANG X X, WANG H J, YANG Z J, LIU X, JIANG W K, MAO L, KONG X Y, JIAO Y N, JIA J Z. Thegenome reveals multiple impacts of transposons. Nature Plants, 2017, 3(12): 946-955.
[31] YU M, MAO S L, HOU D B, CHEN G Y, PU Z E, LI W, LAN X J, JIANG Q T, LIU Y X, DENG M, WEI Y M. Analysis of contributors to grain yield in wheat at the individual quantitative trait locus level. Plant Breeding, 2018, 137(1): 35-49.
[32] R?DER M S, HUANG X Q, B?RNER A. Fine mapping of the region on wheat chromosome 7D controlling grain weight. Functional & Integrative Genomics, 2008, 8(1): 79-86.
[33] HUANG X Q, C?STER H, GANAL M W, R?DER M S. Advanced backcross QTL analysis for the identification of quantitative trait loci alleles from wild relatives of wheat (L.). Theoretical and Applied Genetics, 2003, 106(8): 1379-1389.
[34] MIR R R, KUMAR N, JAISWAL V, GIRDHARWAL N, PRASAD M, BALYAN H S, GUPTA P K. Genetic dissection of grain weight in bread wheat through quantitative trait locus interval and association mapping. Molecular Breeding, 2012, 29(4): 963-972.
[35] CHEN Z Y, CHENG X J, CHAI L L, WANG Z H, BIAN R L, LI J, ZHAO A J, XIN M M, GUO W L, HU Z R, PENG H R, YAO Y Y, SUN Q X, NI Z F. Dissection of genetic factors underlying grain size and fine mapping of QTgw.cau-7D in common wheat (L.). Theoretical and Applied Genetics, 2020, 133(1): 149-162.
[36] LIU H Y, SONG S, XING Y Z. Beyond heading time: FT-like genes and spike development in cereals. Journal of Experimental Botany, 2019, 70(1): 1-3.
[37] 韓領鋒, 亢玲, 張博, 王憲國, 王中華, 張曉科, 陳東升. 小麥光周期基因在我國不同麥區中的組成分布. 麥類作物學報, 2016, 36(12): 1617-1622.
HAN L F, KANG L, ZHANG B, WANG X G, WANG Z H, ZHANG X K, CHEN D S. Composition and distribution of wheat photoperiod genes in different wheat regions in China. Journal of Triticeae Crops, 2016, 36(12): 1617-1622. (in Chinese)
[38] MOCKLER T, YANG H Y, YU X H, PARIKH D, CHENG Y C, DOLAN S, LIN C. Regulation of photoperiodic flowering byphotoreceptors. Proceedings of the National Academy of Sciences of the United States of America, 2003, 100(4): 2140-2145.
[39] WEI J, FANG Y, JIANG H, WU X T, ZUO J H, XIA X C, LI J Q, STICH B, CAO H, LIU Y X. Combining QTL mapping and gene co-expression network analysis for prediction of candidate genes and molecular network related to yield in wheat. BMC Plant Biology, 2022, 22(1): 288.
[40] 呂波. 植物開花基因FT的遺傳轉化及其參與開花調控的研究[D]. 泰安: 山東農業大學, 2014.
Lü B. Genetic transformation of plant flowering gene FT and its involvement in flowering control [D]. Taian: Shandong Agricultural University, 2014. (in Chinese)
[41] LI T, DENG G B, SU Y, YANG Z, TANG Y Y, WANG J H, ZHANG J Y, QIU X, PU X, YANG W Y, LI J, LIU Z H, ZHANG H L, LIANG J J, YU M Q, WEI Y M, LONG H. Genetic dissection of quantitative trait loci for grain size and weight by high-resolution genetic mapping in bread wheat (L.). Theoretical and Applied Genetics, 2022, 135(1): 257-271.
QTL Mapping and Molecular Marker Development of traits related to Grain weight in Wheat
ZHANG ZeYuan1, LI Yue2, ZHAO WenSha1, GU JingJing3, ZHANG AoYan1, ZHANG HaiLong1, SONG PengBo1, WU JianHui1, ZHANG ChuanLiang1, SONG QuanHao4, JIAN JunTao5, SUN DaoJie1, WANG XingRong2
1College of Agronomy, Northwest A&F University, Yangling 712100, Shaanxi;2Institute of Crop Science, Gansu Academy of Agricultural Sciences, Lanzhou 730000;3Luoyang Academy of Agriculture and Forestry Sciences, Luoyang 471023, Henan;4Zhumadian Academy of Agricultural Sciences, Zhumadian 463000, Henan;5Nanyang Academy of Agricultural Sciences, Nanyang 473000, Henan
【Objective】The yield of wheat, the second-highest-yielding food product in the world, has a major impact by grain weight. This research used materials from a recombinant inbred line (RIL) population derived from Heshangtou (HST) and Longchun 23 (LC23). Based on 55K SNP genotype data, QTL mapping was performed for traits related to grain weight of wheat, and co-segregation markers of major grain length QTL were developed and verified to provide reference for molecular marker assisted selection breeding. 【Method】The wheat 55K SNP microarray was used to genotype parents and RIL populations, and a high density genetic linkage map was constructed, and its correlation with Chinese spring reference genome IWGSC RefSeq v1.0 was analyzed.QTL mapping of traits related to grain weight in multiple environments based on inclusive composite interval mapping method.The analysis of variance of major effect QTLs were performed to judge the additive interaction effect among different QTLs, and to analyse its effect on traits related to grain weight.At the same time, the corresponding kompetitive allele specific PCR marker was developed according to the closely linked SNP loci of major QTL for grain length, and verified in 242 wheat accessions worldwide.【Result】In this study, a high density genetic map of Heshangtou/Longchun 23 RIL population was constructed, with full length 4 543 cM, including 22 linkage groups, covering 21 chromosomes of wheat, and the average genetic distance was 1.7 cM.There was a significant correlation between genetic map and physical map, and the Pearson correlation coefficient were 0.77-0.99 (<0.001).A total of 51 QTLs related to grain weight were detected, among them, 4 stable major QTLs were found in multi-environments (three or more environments) and distributed on 2D, 5A, 6B and 7D chromosomes.According to the physical interval and functional markers, it is inferred that stable major QTLsandare photoperiod geneand flowering gene, respectively. The analysis of variance shows that there is a significant interaction between them.The favorite alleles polymerization ofandcan significantly increase thousand grain weight and grain width of wheat.In addition, the corresponding KASP molecular detection markerwas developed based on the co-segregated SNP of the major locusfor grain length,which was significantly correlated with grain length and grain weight traits in a diversity panel comprising of 242 wheat accessions, and could increase grain length by 3.33% to 4.59% and grain weight 5.70% to 10.35% in different environments (<0.001). 【Conclusion】There are several genetic loci that affect traits linked to grain weight in Heshangtou (HST) and Longchun 23 (LC23), andanddramatically increased thousand grain weight and grain width through additive interaction effects.is significantly correlated with grain weight and grain length, and its co-segregated molecular markercan be used in molecular marker assisted selection breeding.
wheat; thousand-grain weight; QTL; KASP marker; molecular marker-assisted selection breeding

2023-03-28;
2023-04-20
陜西省“兩鏈”融合重點專項(2023KXJ-011)
張澤源,E-mail:18238768351@163.com。通信作者孫道杰,E-mail:sunwheat@nwsuaf.edu.cn。通信作者王興榮,E-mail:wxr_0618@163.com
(責任編輯 李莉)
