副溶血弧菌QsvR ChIP-qPCR方法的建立
2023-11-24 09:15:38張苗苗李雪陸仁飛張義全周敏
江蘇大學學報(醫學版) 2023年6期
關鍵詞:實驗
張苗苗, 李雪, 陸仁飛, 張義全, 周敏
(1. 南通大學附屬南通第三醫院檢驗科, 江蘇 南通 226006; 2. 江蘇大學醫學院, 江蘇 鎮江 212013; 3. 南通市疾病預防控制中心微生物科, 江蘇 南通 226007)
在轉錄調控機制研究中,通常需要測定調控蛋白質是否能與靶基因啟動子區DNA序列相互作用并確定對應的作用位點,常用的實驗方法為凝膠阻滯實驗(electrophoretic mobility shift assays,EMSA)和DNase I足跡實驗[1]。然而,EMSA和DNase I足跡實驗均屬于體外實驗,不能準確反映蛋白質在細菌體內的準確結合序列,而且常出現假陽性結果。此外,DNase I足跡實驗還需要特殊的設備(如測序儀)或原材料(如放射性同位素),不適合作為實驗室的常規實驗方法。因此,急需要一種替代實驗方法來解決上述問題。
染色質免疫共沉淀技術(chromatin immuno-precipitation,ChIP)是利用特異性抗原抗體反應,在基因組水平上研究生命體組織或者細胞內蛋白質與DNA相互作用的一種技術方法[2]。ChIP技術在真核生物中已經得到廣泛的應用,具有多種相應的試劑盒,但是這些試劑盒并不適用于原核生物[3]。
副溶血弧菌(Vibrioparahaemolyticus)是一種革蘭陰性嗜鹽性弧菌,廣泛存在于近海海水、海底沉淀物以及海產品中,感染后可引起以腹瀉、腹痛、低燒、嘔吐等為主要癥狀的急性腸胃炎[4]。副溶血弧菌可分泌多種毒力因子包括耐熱直接溶血素(thermostable direct hemolysin,TDH)、耐熱直接溶血素相關溶血素(TDH-related hemolysin,TRH)、Ⅲ型分泌系統(T3SS)和Ⅵ型分泌系統(T6SS)等[5-6]。AphA和OpaR分別是副溶血弧菌密度感應系統在低密度和高密度下的核心調控子,參與調控生物膜形成、毒力因子、運動等生命活動[7-9]。QsvR是AraC家族的轉錄調控子,與密度感應系統協同調控毒力基因的表達[1]。在低密度條件下,AphA間接抑制qsvR的轉錄;高密度時,OpaR間接激活qsvR的轉錄[1]。QsvR在高密度條件下表達量較高,主要在高密度條件下發揮調控作用,直接抑制和激活aphA和opaR的轉錄[1]。QsvR直接激活T3SS1相關基因exsBAD-vscBCD,T3SS2相關基因vtrA(vpa1332-1333)、vopB2(vpa1362-1358)和tdh2,T6SS2相關基因vpa1027-1024、vpa1043-1028和vpa1044-1046的轉錄[1,10]。此外,QsvR抑制相關極鞭毛基因的表達,而對側鞭毛基因無調控作用,對生物膜的形成也具有一定的調控作用[11-12]。
本研究旨在建立副溶血弧菌中ChIP-qPCR的方法,為后續研究原核生物體內蛋白質和DNA序列相互作用的調控通路提供可靠的實驗手段。
1 材料與方法
1.1 材料
1.1.1 菌株 副溶血弧菌RIMD2210633qsvR非極性突變株(ΔqsvR)、大腸埃希菌S17 λpir和重組質粒pBAD33-qsvR-2×Flag由南通大學附屬南通第三醫院檢驗科保存。
1.1.2 主要試劑 HI培養基(2.5%腦心浸肉湯培養基)購自美國BD Bioscience公司;抗Flag M2磁珠購自美國SigmaAldrich公司;即用型溶菌酶購自美國Epicentre Biotechnologies公司;蛋白酶抑制劑(PIC)購自瑞士Roche Applied Sciences公司;SuperReal熒光定量預混試劑彩色版(SYBR Green)和FastKing一步法除基因組cDNA第一鏈合成預混試劑購自天根生化科技(北京)有限公司;質粒提取試劑盒和PCR產物純化試劑盒購自QIAGEN公司;TRIzol試劑購自美國Invitrogen公司;抗Flag標簽單克隆抗體和辣根過氧化物酶(HRP)標記的二抗購自索萊寶生物科技有限公司。
1.2 pBAD33-qsvR-2×Flag重組質粒的熱激轉化
取100 μL大腸埃希菌S17 λpir感受態細胞于冰上融化,加入100 ng pBAD33-qsvR-2×Flag重組質粒,冰上孵育30 min。42 ℃、90 s進行熱休克,立即冰浴2 min,加入900 μL事先預熱的LB液體培養基(1% NaCl、1%胰蛋白胨和0.5%酵母粉),于37 ℃靜置孵育1 h。4 ℃、4 000 r/min離心5 min,棄去900 μL上清液,將剩下100 μL菌液混勻,均勻涂布于含20 μg/mL氯霉素的LB平板(1% NaCl、1%胰蛋白胨、0.5%酵母粉和1.5%瓊脂)上,37 ℃培養出單克隆。選取3個單克隆以qsvR-RT-F/R和pBAD33-F/R為引物(表1),進行PCR鑒定[13],此時菌株記為S17/pBAD33-qsvR-2×Flag。

表1 所用引物序列
1.3 pBAD33-qsvR-2×Flag重組質粒的接合轉移
取10 μL S17/pBAD33-qsvR-2×Flag和副溶血弧菌ΔqsvR甘油菌株分別接種至5 mL LB液體培養基中,37 ℃、200 r/min,培養12~14 h。分別取500 μL S17/pBAD33-qsvR-2×Flag和ΔqsvR,4 000 r/min離心5 min收集菌體,然后用1 mL無菌磷酸鹽緩沖液(PBS)洗滌2次,每次4 000 r/min離心3 min。分別用100 μL LB液體培養基重懸菌體,并混合在一起,取100 μL混合菌液點種在LB平板上。待菌液干燥后,將LB平板30 ℃靜置培養6~8 h。用1 mL無菌PBS刮取LB平板上的菌斑,取100 μL涂布于含有5 μg/mL氯霉素和100 μg/mL氨芐西林的LB平板上,37 ℃靜置培養至分離出單克隆。選取3個單克隆以toxR-RT-F/R、qsvR-RT-F/R和pBAD33-F/R為鑒定引物(表1),作PCR鑒定[13],此時菌株記為ΔqsvR/pBAD33-qsvR-2×Flag。
1.4 細菌培養
取10 μLΔqsvR/pBAD33-qsvR-2×Flag甘油菌種接種于5 mL的HI肉湯中,37 ℃、200 r/min培養12 h。50倍稀釋轉接至5 mL HI肉湯中,37 ℃、200 r/min培養至D(600 nm)為1.2~1.4。1 000倍稀釋轉接至50 mL HI肉湯中,設置加阿拉伯糖誘導組(Ara+),未加阿拉伯糖誘導(Ara-)作為對照,37 ℃、200 r/min培養至D(600 nm)為1.0。氯霉素工作濃度為5 μg/mL,阿拉伯糖為0.1%。
1.5 蛋白質印跡實驗檢測QsvR-2×Flag蛋白
ΔqsvR/pBAD33-qsvR-2×Flag菌株按照“1.4”方法培養后,分別取加阿拉伯糖和不加阿拉伯糖誘導的5 mL菌液,用PBS緩沖液懸起菌體并超聲碎菌,4 ℃、12 000 r/mim離心10 min后,取上清液,用Micro BCATMProtein Assay Kit測定上清液中的總蛋白含量。取等量總蛋白的阿拉伯糖誘導和未誘導的裂解上清液進行10% SDS-PAGE電泳,而后轉移至PVDF膜上,以抗Flag標簽單克隆抗體為一抗,HRP標記的IgG為二抗,進行蛋白質印跡實驗[14],通過比較特異性條帶的差異來判斷QsvR-2×Flag的表達量。
1.6 實時定量PCR檢測qsvR mRNA的表達
ΔqsvR/pBAD33-qsvR-2×Flag菌株按照“1.4”方法培養后,采用TRIzol法提取阿拉伯糖誘導和未誘導菌體的總RNA。分別取1 μg的總RNA,利用FastKing一步法除基因組cDNA第一鏈合成預混試劑盒將其逆轉錄成cDNA,然后用SuperReal熒光定量預混試劑彩色版(SYBR Green)通過實時熒光定量PCR儀進行qPCR分析[15]。以16S rRNA基因作為內參,采用2-ΔΔCt法對qsvR的表達水平進行相對定量,所用引物見表1。
1.7 DNA和蛋白質的交聯
ΔqsvR/pBAD33-qsvR-2×Flag菌株按照“1.4”方法培養后,分別取阿拉伯糖誘導和未誘導的菌液40 mL,添加1.1 mL 37%的甲醛溶液,置30 ℃、100 r/min下作用10 min。10 mL/管分裝至無核酸酶離心管中,向每管中添加2.5 mol/L的甘氨酸溶液1 mL,4 ℃旋轉孵育30 min。4 ℃、4 649×g離心10 min,沉淀用10 mL預冷的PBS洗滌2次,重懸于1 mL預冷的PBS中,隨后補加25×蛋白酶抑制劑40 μL和100 mmol/L苯基甲基磺酰氟10 μL,混勻。4 ℃、16 000×g離心5 min,棄上清液,沉淀保存至-80 ℃冰箱中。
1.8 超聲裂菌
分別取一管加阿拉伯糖誘導和未誘導的交聯后菌體,用0.5 mL裂解液(10 mmol/L Tris-HCl、50 mmol/L NaCl、EDTA、14.5 kU/mL溶菌酶和20 μg/mL RNase A)重懸,37 ℃孵育30 min。加入0.5 mL 2×IP緩沖液(200 mmol/L Tris-HCl、600 mmol/L NaCl、4% Triton X-100、0.2×蛋白酶抑制劑和2 mmol/L苯基甲基磺酰氟),渦旋混勻。100 W超聲裂菌,裂菌參數為超聲裂解6 s、間隔6 s,共9 min。4 ℃、16 000×g離心10 min,每管上清液200 μL分裝至無核酸酶EP管中。分別取出一管超聲上清液添加4 μL 5 mol/L NaCl,65 ℃孵育4 h,充分渦旋混勻后,16 000×g離心3 min。將上層水相利用酚氯仿法提取總DNA,進行1%瓊脂糖凝膠電泳分析,DNA片段應均在100~1 000 bp之間。
1.9 ChIP反應
分別取阿拉伯糖誘導和未誘導的超聲裂解液上清液0.4 mL,其中0.2 mL用作Input樣品,0.2 mL用作IP。用裂解液將IP樣品補加到1 mL,加入到50 μL經平衡的抗Flag磁珠中,4 ℃旋轉孵育過夜。用1 mL洗滌液1(100 mmol/L Tris-HCl pH 7.5、250 mmol/L LiCl和2% Triton X-100)、洗滌液2(100 mmol/L Tris-HCl pH 7.5、600 mmol/L NaCl和2% Triton X-100)和洗滌液3(100 mmol/L Tris-HCl pH 7.5、300 mmol/L NaCl和2%Triton X-100)分別洗滌3次,再用TE洗滌液(10 mmol/L Tris-HCl pH 8.0和1 mmol/L EDTA)洗滌3次。將磁珠重懸于0.2 mL洗脫液(10 mmol/L Tris-HCl pH 8.0、1 mmol/L EDTA和1%十二烷基硫酸鈉)中。洗脫產物于65 ℃孵育2 h,3 100×g離心30 s,上清液轉移至無核酸酶EP管中。
1.10 去交聯和qPCR
向Input和IP DNA中加入5 mol/L NaCl 8 μL,65 ℃孵育4 h,以去交聯。加入0.5 mol/L EDTA 5 μL、10 μL 1 mol/L Tris-HCl pH 7.0和10 mg/mL蛋白酶K 2 μL,45 ℃孵育1 h。用PCR產物純化試劑盒純化Input和IP樣品,用0.1~0.2 mL的無菌去離子水洗脫DNA。參考文獻[16]的方法,實時定量PCR檢測目標DNA(aphA、opaR、exsB和vtrA)的IP量,計算公式為2(Ct Input-Ct IP)。
1.11 統計學方法
應用GraphPad Prism 9.4.1和Visio Professional 2019進行相關統計學分析并進行繪圖。qPCR實驗至少形成3次獨立實驗,每次實驗3個生物學重復,結果用均值±標準差表示,采用雙尾t檢驗進行組間比較,P<0.05為差異有統計學意義。
2 結果
2.1 pBAD33-qsvR-2×Flag重組質粒的鑒定
將pBAD33-qsvR-2×Flag重組質粒熱激轉化進入大腸埃希菌S17 λpir,PCR鑒定結果見圖1,相較于pBAD33空質粒,陽性克隆的pBAD33質粒中克隆入了qsvR基因片段,分子量較大,電泳條帶滯后。
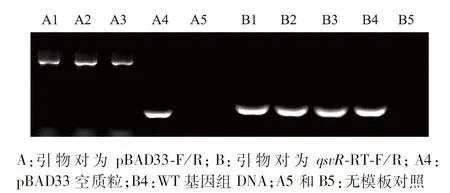
A:引物對為pBAD33-F/R;B:引物對為qsvR-RT-F/R;A4:pBAD33空質粒;B4:WT基因組DNA;A5和B5:無模板對照
2.2 ChIP-qPCR實驗菌株的鑒定
將pBAD33-qsvR-2×Flag重組質粒結合轉移入ΔqsvR中,獲得ΔqsvR/pBAD33-qsvR-2×Flag,PCR驗證后,保存菌種用作實驗菌株。提取ΔqsvR/pBAD33-qsvR-2×Flag的總蛋白,蛋白質印跡實驗顯示,未加阿拉伯糖誘導的菌株中QsvR-2×Flag蛋白的表達低于檢測下線,而經阿拉伯糖誘導的菌株QsvR-2×Flag蛋白大量表達。提取ΔqsvR/pBAD33-qsvR-2×Flag的總RNA,qPCR結果可見,經阿拉伯糖誘導的菌株中qsvR的mRNA豐度是未加阿拉伯糖的130多倍。見圖2。由此可見,阿拉伯糖誘導與否qsvR的表達量存在顯著差異,可用無阿拉伯糖誘導的菌株ΔqsvR/pBAD33-qsvR-2×Flag作為本研究的對照組。
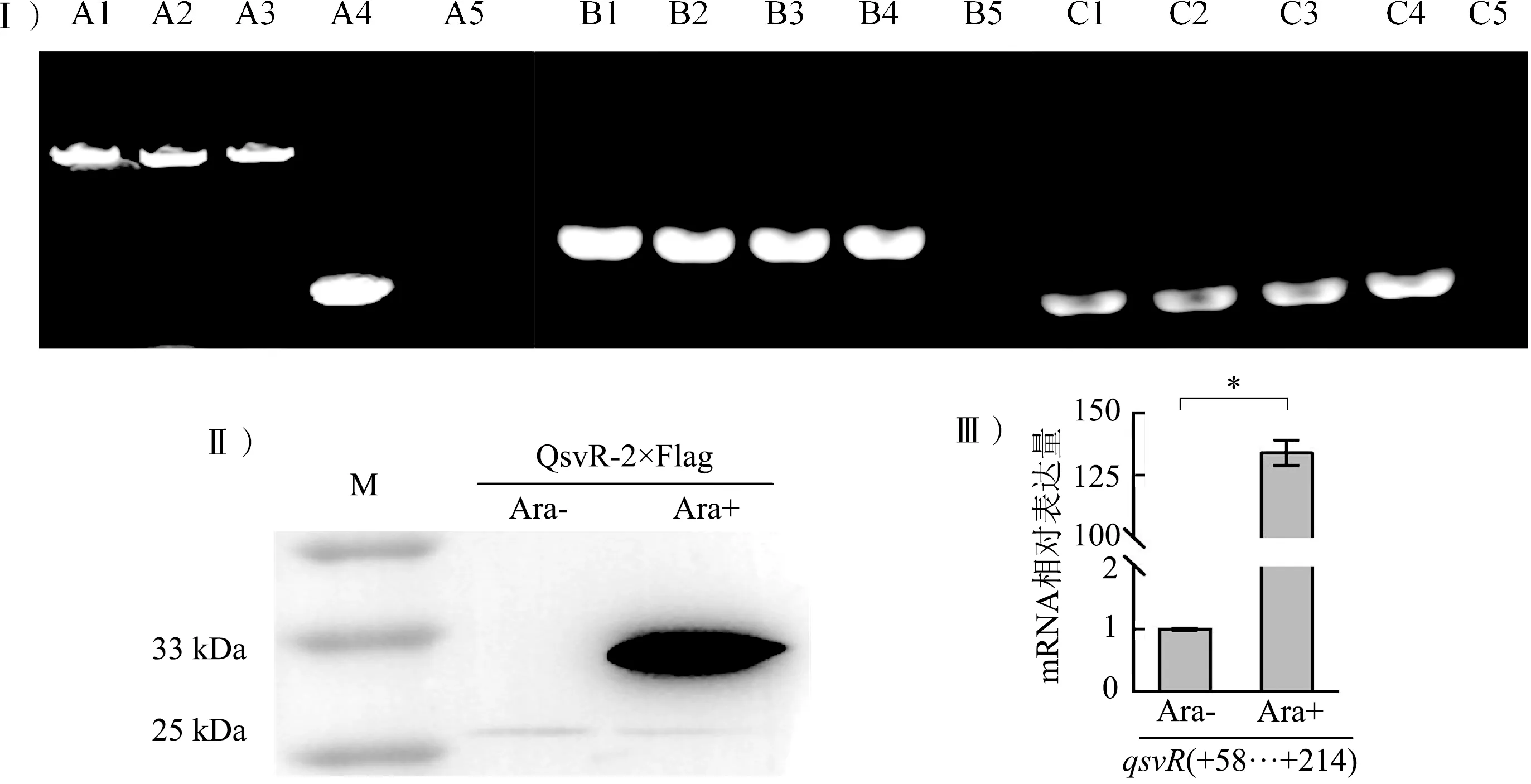
Ⅰ:PCR驗證實驗菌株,A:引物對為pBAD33-F/R;B:引物對為qsvR-RT-F/R;C:引物對為toxR-RT-F/R;A4:pBAD33空質粒;B4和C4:WT基因組DNA;A5、B5和C5:無模板對照。Ⅱ:蛋白質印跡。Ⅲ:qPCR,*:P<0.05
2.3 副溶血弧菌中QsvR的ChIP-qPCR
將pBAD33-qsvR-2×Flag重組質粒轉入ΔqsvR中,經阿拉伯糖誘導后表達QsvR-2×Flag蛋白,在副溶血弧菌中可以與基因組DNA交聯形成2×Flag-QsvR-DNA復合體,通過抗原抗體特異性反應將蛋白質-DNA復合體沉淀出來,解交聯后獲得與QsvR直接結合的DNA,利用qPCR分析可獲得QsvR與DNA相互作用的信息。結果如圖3所示,相較于未經阿拉伯糖誘導的菌株,經阿拉伯糖誘導菌株的aphA、opaR、exsB和vtrA的IP量明顯較高,分別高3.68、8.02、9.44和8.05倍,表明在副溶血弧菌體內QsvR與aphA、opaR、exsB和vtrA具有結合作用。對于16S rRNA,二者之間沒有明顯的差異,表明QsvR與16S rRNA不結合。

*:P<0.05
3 討論
ChIP是在活細胞狀態下分析蛋白質與DNA相互作用的一種實驗技術,可以彌補常用實驗方法的不足。其原理是,靶蛋白質氨基酸序列末端標記特定標簽,在菌體內與DNA進行交聯,形成蛋白質-DNA復合物;提取菌體基因組DNA并將其裂解為100~1 000 bp大小不等的片段,經特定單克隆抗體與蛋白質中的標簽進行特異性抗原抗體反應沉淀蛋白質-DNA復合物,解交聯后收集DNA,利用qPCR檢測蛋白質結合靶DNA的量[3]。ChIP不僅可以與qPCR相結合分析轉錄調控子與特定靶基因的相互作用信息,也可以結合測序技術來探究蛋白質的未知靶基因或者識別特定蛋白質在整個基因組中的所有靶基因[3]。目前,在原核生物中,已有ChIP應用的案例,例如,利用ChIP-qPCR確定了在大腸埃希菌體內與σ因子RpoE直接相互作用的靶基因[16];ChIP與測序技術相結合分析出溶藻弧菌(V.alginolyticus)基因組內DNA結合蛋白ToxR和LuxR的結合位點[17-18]。本研究對交聯、免疫沉淀反應和解交聯等步驟進行了優化,以副溶血弧菌中調控子蛋白QsvR為研究對象,成功建立了ChIP-qPCR實驗技術,并驗證了其準確性和穩定性。
QsvR是AraC家族的DNA結合蛋白,在副溶血弧菌中與密度感應系統的兩個核心調控子AphA和OpaR協同調控毒力和生物膜相關基因的轉錄[1,11]。體外的凝膠阻滯實驗和DNase Ⅰ足跡實驗表明QsvR與aphA(-312…-175)、opaR(-190…-72)、exsB(-200…-88)和vtrA(-148…-67)啟動子區DNA片段具有結合作用,與16S rRNA片段無結合作用[1]。本研究建立了副溶血弧菌中的ChIP-qPCR,并且驗證了體外實驗的結果,經阿拉伯糖誘導的菌株aphA、opaR、exsB和vtrA的IP量比未誘導菌株明顯升高,表明QsvR與aphA、opaR、exsB和vtrA具有直接的結合作用,解決了沒有相應試劑盒的困境,為后續研究原核生物體內調控通路提供了可靠的實驗手段。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
