地黃苷A通過調節AKT/Nrf2/GPX4信號通路介導的鐵死亡改善大鼠心肌缺血再灌注損傷
2023-11-25 08:37:02王曉英
中西醫結合心腦血管病雜志 2023年22期
劉 盼,王曉英,韓 丹
冠心病具有高發病率和高死亡率等特點,心肌缺血再灌注損傷(myocardial ischemia reperfusion injury,MI/RI)是冠心病的主要病理表現,主動脈血管壁斑塊破裂可促進血栓形成并阻塞血管腔,造成心肌缺血性損傷,在一定時間內恢復血液供應,導致心肌二次受損,這一過程被稱為MI/RI[1-2]。MI/RI發病機制復雜,其中氧化應激、細胞凋亡、炎癥等因素均參與MI/RI發生發展[3]。近年來研究發現,鐵死亡是導致MI/RI發展的重要因素[4]。鐵死亡是一種新的細胞死亡方式,具有鐵離子依賴性,鐵離子在細胞內代謝失衡可增加細胞內鐵離子的蓄積,進而催化細胞膜不飽和脂肪酸發生脂質過氧化反應,誘導細胞鐵死亡[5]。研究發現,蛋白激酶B(protein kinase B,AKT)/核因子紅細胞系2相關因子2(nuclear factor erythroid 2-related factor 2,Nrf2)/谷胱甘肽過氧化酶4(glutathione peroxidase 4,GPX4)信號通路的激活可抑制鐵死亡[6-7]。地黃苷A(Rehmannioside A,ReA)是玄參科植物地黃中的重要活性成分,具有抗氧化、抗炎、抗凋亡等療效[8]。研究發現,ReA可通過激活AKT/Nrf2/GPX4信號通路,抑制神經元鐵死亡,從而發揮神經保護作用[9]。而ReA能否通過調控AKT/Nrf2/GPX4信號通路介導的鐵死亡改善MI/RI尚鮮見報道。為此,本研究以鐵死亡為切入點,旨在探究ReA對MI/RI大鼠的影響及其作用機制。
1 材料與方法
1.1 實驗動物
90只體質量為220~250 g的無特定病原體(SPF)級SD大鼠,購自湖北貝恩特生物科技有限公司,生產許可證號為SCXK(鄂)2021-0027。所有大鼠均飼喂于溫度24 ℃和濕度50%的動物房中,并提供12 h光/暗循環。
1.2 主要試劑
ReA(貨號:YT61748)購自北京伊塔生物科技有限公司;Ferrostatin-1(Fer-1)(貨號:F27030)購自上海吉至生化科技有限公司;perifosine(貨號:S43190)購自上海源葉生物科技有限公司;乳酸脫氫酶(LDH)(貨號:E-EL-R0576)、肌酸激酶同工酶MB(CK-MB)(貨號:E-EL-R1327)和心肌肌鈣蛋白I(cTnI)(貨號:E-EL-R1253)檢測試劑盒均購自上海恒斐生物科技有限公司;氯代三苯基四氮唑(TTC)(貨號:S0022)購自上海遠慕生物科技有限公司;蘇木素-伊紅(HE)染色試劑盒(貨號:C0105)和原位末端標記測定法(TUNEL)染色試劑盒(貨號:C1086)均購自上海碧云天生物技術有限公司;谷胱甘肽過氧化物酶(GSH-Px)(貨號:BC1190)、超氧化物歧化酶(SOD)(貨號:BC0170)和丙二醛(MDA)(貨號:BC0020)檢測試劑盒均購自北京索萊寶科技有限公司;Fe2+含量檢測試劑盒(貨號:E1050)購自北京普利萊基因技術有限公司;兔源p-AKT(貨號:ab192623)、AKT(貨號:ab179463)、Nrf2(貨號:b92946)、GPX4(貨號:ab125066)、轉鐵蛋白受體1(TfR1)(貨號:ab269513)、GAPDH(貨號:ab199554)和辣根過氧化物酶(HRP)標記的山羊抗兔二抗(貨號:ab205718)均購自Abcam公司。
1.3 動物分組及MI/RI大鼠模型建立
90只大鼠適應性喂養1周后,隨機分為假手術組(Sham組)、模型組(Model組)、ReA低劑量組(ReA-L組)、ReA高劑量組(ReA-H組)、鐵死亡抑制劑Fer-1組(Fer-1組)及ReA高劑量+AKT抑制劑perifosine組(ReA-H+perifosine組),每組15只。ReA-L組、ReA高劑量組大鼠分別腹腔注射40、80 mg/kg ReA[10],Fer-1組大鼠腹腔注射2 mg/kg ferrostatin-1[11],ReA-H+perifosine組大鼠腹腔注射80 mg/kg ReA及20 mg/kg perifosine[12],Sham組和Model組大鼠腹腔注射等量生理鹽水,每日1次,連續給藥2周。除Sham組外,各組大鼠在末次給藥1 h后建立MI/RI大鼠模型,參照文獻[13]方法,應用左冠狀動脈前降支結扎法:將大鼠用1%戊巴比妥鈉(30 mg/kg)腹腔注射麻醉后,以仰臥位姿勢固定在手術臺上;胸部皮膚經備皮消毒后,在大鼠左側第3肋與第4肋骨間行開胸手術,暴露心臟;在冠狀動脈左前降支進行30 min結扎,此時可觀察到心肌由紅色變為白色;30 min后打開結扎線再灌注2 h,心肌由白色變為紅色,提示建模成功。Sham組大鼠除不進行冠狀動脈結扎外,其余操作同建模大鼠一致。
1.4 血清心肌酶水平檢測
造模結束后,采集各組大鼠腹主動脈血液5 mL離心獲得血清;按LDH、CK-MB和cTnI酶聯免疫試劑盒要求檢測大鼠血清LDH、CK-MB和cTnI的水平。
1.5 心肌梗死面積檢測
每組取5只大鼠經頸椎脫臼處死,采集左心室組織;使用切片刀將左心室切割為5片,每片2 mm厚;將切割完成的心肌組織置于1% TTC溶液中進行染色;隨后轉移到10%甲醛溶液中固定;正常心肌組織呈紅色,梗死區心肌組織呈白色。采用Image J計算梗死面積,梗死面積為每片切片梗死心肌面積總和占左心室面積的百分比。
1.6 心肌組織病理形態學觀察
每組再取5只大鼠經頸椎脫臼處死后,采集左心室缺血區心肌組織固定于4%多聚甲醛中;固定24 h后,心肌組織依次經脫水、透明、包埋后,采用連續切片機進行5 μm連續切片;依次將切片置入蘇木素染色液和伊紅染色液中進行染色;光學顯微鏡下觀察心肌組織染色結果。
1.7 心肌細胞凋亡率檢測
取心肌組織切片經脫蠟至水后,再切片添加蛋白酶K工作液孵育;滴加破膜工作液破膜;將TUNEL試劑盒內的TDT酶、dUTP和buffer按1∶5∶50比例混合配制反應液;滴加反應液到切片孵育;DAPI染液復染細胞核;熒光顯微鏡下觀察染色結果。
1.8 心肌組織抗氧化能力評估
每組另取5只大鼠經頸椎脫臼處死后,采集左心室缺血區心肌組織置于勻漿管中進行勻漿;按SOD、GSH-Px和MDA酶聯免疫試劑盒要求評估心肌組織抗氧化能力。
1.9 心肌組織Fe2+含量檢測
取心肌組織勻漿,加入Fe2+檢測試劑,具體操作依照Fe2+含量檢測試劑盒說明書進行,然后在550 nm波長處測定吸光度值,并按照標準曲線計算Fe2+含量。
1.10 心肌組織蛋白表達檢測
取心肌組織勻漿,在勻漿液中加入RAPI裂解液離心后獲得總蛋白;使用二喹啉甲酸法(BCA)蛋白檢測試劑盒檢測其濃度;然后行SDS-PAGE、轉膜及封膜處理;將p-AKT(1∶1 000)、AKT(1∶1 000)、Nrf2(1∶500)、GPX4(1∶1 000)、TfR1(1∶1 000)和GAPDH(1∶1 000)一抗加入膜中并在室溫下孵育過夜;將HRP標記的二抗加入膜孵育;采用Image LabTM軟件分析蛋白的灰度值。
1.11 統計學處理
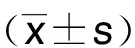
2 結 果
2.1 各組大鼠血清心肌酶水平比較
與Sham組比較,Model組大鼠血清LDH、CK-MB和cTnI水平升高(P<0.05);與Model組比較,ReA-L組、ReA-H組、Fer-1組大鼠血清LDH、CK-MB和cTnI水平降低,且隨ReA劑量增加,大鼠血清LDH、CK-MB和cTnI水平進一步降低(P<0.05);與ReA-H組比較,ReA-H+perifosine組大鼠血清LDH、CK-MB和cTnI水平升高(P<0.05)。詳見表1。

表1 各組大鼠血清LDH、CK-MB和cTnI水平比較
2.2 各組大鼠心肌梗死面積比較
Model組大鼠的心肌梗死面積高于Sham組(P<0.05);與Model組相比,ReA-L組、ReA-H組、Fer-1組大鼠心肌梗死面積降低,且隨著ReA劑量增加,大鼠心肌梗死面積進一步減少(P<0.05);與ReA-H組比較,ReA-H+perifosine組大鼠心肌梗死面積升高(P<0.05)。詳見表2、圖1。

圖1 TTC觀察大鼠心肌梗死面積
表2 各組大鼠心肌梗死面積比較
2.3 各組大鼠心肌組織病理學損傷比較
Model組與Sham組比較,大鼠心肌組織明顯受損,心肌細胞腫脹且雜亂排列,心肌纖維紊亂,有大量炎性細胞浸潤;ReA-L組、ReA-H組、Fer-1組大鼠心肌組織損傷明顯減輕,且隨ReA劑量增加,效果越明顯;與ReA-H組相比,ReA-H+perifosine組大鼠心肌組織損傷明顯加重。詳見圖2。

圖2 大鼠心肌組織病理形態學變化(HE染色,×400)
2.4 各組大鼠心肌細胞凋亡情況比較
Model組與Sham組比較,心肌細胞凋亡率升高(P<0.05);與Model組比較,ReA-L組、ReA-H組、Fer-1組心肌細胞凋亡率降低(P<0.05);ReA-H+perifosine組與ReA-H組相比,心肌細胞凋亡率升高(P<0.05)。詳見圖3、表3。

圖3 TUNEL染色檢測大鼠心肌細胞凋亡率變化(×200)
表3 各組大鼠心肌細胞凋亡率比較
2.5 各組大鼠心肌組織抗氧化指標水平比較
Model組與Sham組相比,心肌組織GSH-Px和SOD水平降低,MDA水平升高(P<0.05);ReA-L組、ReA-H組、Fer-1組與Model組比較,心肌組織GSH-Px和SOD水平升高,MDA水平下降,ReA各劑量組間GSH-Px、SOD、MDA水平差異有統計學意義(P<0.05);與ReA-H組比較,ReA-H+perifosine組GSH-Px和SOD水平下降,MDA水平增高(P<0.05)。詳見表4。
表4 各組大鼠心肌組織GSH-Px、SOD和MDA水平比較
2.6 各組大鼠心肌組織Fe2+含量比較
Model組與Sham組比較,Fe2+含量增加(P<0.05);ReA-L組、ReA-H組、Fer-1組與Model組比較,Fe2+含量降低,且ReA各劑量組間Fe2+含量差異有統計學意義(P<0.05);與ReA-H組比較,ReA-H+perifosine組大鼠心肌組織Fe2+含量升高(P<0.05)。詳見表5。
表5 各組大鼠心肌組織Fe2+含量比較
2.7 各組大鼠心肌組織蛋白p-AKT/AKT、Nrf2、GPX4和TfR1表達比較
Model組與Sham組比較,p-AKT/AKT、Nrf2和GPX4蛋白水平下調,TfR1蛋白水平升高(P<0.05);與Model組比較,ReA-L組、ReA-H組、Fer-1組心肌組織p-AKT/AKT、Nrf2和GPX4蛋白水平升高,TfR1蛋白水平下調,隨ReA劑量增加,大鼠心肌組織p-AKT/AKT、Nrf2和GPX4蛋白水平進一步升高,TfR1蛋白水平進一步降低(P<0.05);與ReA-H組比較,ReA-H+perifosine組大鼠心肌組織p-AKT/AKT、Nrf2和GPX4蛋白表達降低,TfR1蛋白表達增高(P<0.05)。詳見圖4、表6。

圖4 Western Blot檢測各組大鼠心肌組織p-AKT/AKT、Nrf2、GPX4和TfR1蛋白表達條帶圖
表6 各組大鼠心肌組織p-AKT/AKT、Nrf2、GPX4和TfR1蛋白表達比較
3 討 論
MI/RI是心肌缺血恢復血液供應后心肌損傷反而加重的一種現象,是冠心病的主要病理表現[14]。伴隨著MI/RI發展可造成急性心肌梗死的發生,而急性心肌梗死是導致冠心病病人死亡的主要原因[15]。控制MI/RI發生發展是改善冠心病病人預后的關鍵。MI/RI發病機制復雜尚未完全闡明,因此,闡明MI/RI發病機制對控制MI/RI發展具有重要意義。本研究采用左冠狀動脈前降支結扎法建立MI/RI大鼠模型,結果顯示,與Sham組比較,Model組大鼠心肌梗死面積及心肌細胞凋亡率增加,心肌組織形態結構嚴重受損;此外,血清中心肌酶LDH、CK-MB和cTnI水平上升,這可能是大量心肌細胞損傷,導致胞內心肌酶釋放到血液中的結果,以上結果進一步提示了MI/RI大鼠模型成功建立。研究發現,MI/RI發生后,心肌細胞中往往存在大量鐵離子蓄積,進而促進細胞脂質過氧化發生,誘導細胞鐵死亡,加重組織損傷[16]。在本研究中,與Sham組相比,Model組大鼠心肌組織中Fe2+含量增加,且心肌組織中GSH-Px和SOD水平降低,MDA水平升高,提示了鐵死亡與MI/RI發生密切相關。
當前,尚無十分有效的治療方法來保護心臟免受MI/RI,因此,尋求及開發新策略來預防MI/RI具有重要的臨床意義。ReA來源于玄參科植物地黃的干燥塊根,已被證實具有抗氧化、抗炎、抗凋亡等生物學功能[17]。研究報道,ReA可提高認知障礙模型大鼠腦組織中GSH-Px和內皮型一氧化氮合成酶(eNOS)水平,降低MDA產生,進而抑制脂質過氧化、細胞凋亡和細胞鐵死亡的發生[9]。在本研究中,低、高劑量ReA及Fer-1處理后大鼠心肌組織中Fe2+含量及MDA水平降低,GSH-Px和SOD水平上升,且血清中心肌酶LDH、CK-MB和cTnI水平下降,心肌梗死面積及心肌細胞凋亡率降低,心肌組織損傷減輕,提示ReA可能通過抑制心肌細胞鐵死亡,改善心肌組織損傷。
細胞鐵死亡機制與細胞內鐵離子代謝失衡及氧化損傷密切相關[18]。TfR1是調控細胞內鐵離子代謝的重要蛋白,負責循環中Fe3+跨膜轉運到細胞內被還原為Fe2+所利用,研究發現,敲低TfR1可通過抑制細胞鐵死亡改善MI/RI[19]。細胞發生鐵死亡時Fe2+可在細胞內蓄積,因Fe2+具有強氧化性,可與細胞內H2O2反應引起大量羥自由基生成,促進脂質過氧化發生,進而損傷細胞膜[20]。GPX4是調節細胞鐵死亡的關鍵蛋白,GPX4表達降低可打破細胞內氧化平衡,誘導脂質過氧化發生,進而促進細胞鐵死亡[21]。Nrf2受AKT的調控,是維持細胞氧化還原穩態的關鍵轉錄因子,AKT-Nrf2通路的激活可促使下游GPX4表達升高,抑制細胞鐵死亡的發生[7]。本研究結果顯示,Model組大鼠心肌組織p-AKT/AKT、Nrf2和GPX4蛋白水平下調,TfR1蛋白水平升高,而經低、高劑量ReA及Fer-1處理后心肌組織p-AKT/AKT、Nrf2和GPX4蛋白水平上升,TfR1蛋白水平下降,提示ReA可能通過激活AKT/Nrf2/GPX4信號通路抑制心肌細胞鐵死亡,改善心肌組織損傷。為驗證ReA是否通過激活AKT/Nrf2/GPX4信號通路,抑制心肌細胞鐵死亡,本研究采用AKT抑制劑perifosine處理干預高劑量ReA大鼠,結果顯示,與ReA-H組比較,ReA-H+perifosine組大鼠心肌組織p-AKT/AKT、Nrf2和GPX4蛋白水平降低,心肌細胞內Fe2+含量增高,心肌組織損傷嚴重,以上結果提示perifosine減弱了高劑量ReA對MI/RI大鼠鐵死亡的抑制作用,證實了ReA能夠通過抑制心肌細胞鐵死亡進而改善心肌組織損傷,這可能與AKT/Nrf2/GPX4信號通路的激活有關。
綜上所述,ReA能夠抑制心肌細胞鐵死亡,進而改善MI/RI,這可能是通過激活AKT/Nrf2/GPX4信號通路實現的。本研究為開發預防和治療MI/RI新藥提供了理論依據。然而,ReA對MI/RI發生后心肌組織的保護作用可能還涉及其他通路,仍需進一步探究。
猜你喜歡
課堂內外·初中版(科學少年)(2023年10期)2023-12-10 00:43:06
全科護理(2022年10期)2022-12-26 21:19:15
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
鄉村科技(2021年33期)2021-03-16 02:26:54
國際放射醫學核醫學雜志(2021年10期)2021-02-28 08:41:58
火花(2019年12期)2019-12-26 01:00:28
人大建設(2019年12期)2019-05-21 02:55:32
學苑創造·A版(2015年11期)2016-01-14 09:03:27
中國合理用藥探索(2014年11期)2014-03-11 20:30:20
