板青敗毒口服液的HPLC特征圖譜研究
2023-12-01 05:24:00張傳津牛華星韓會剛戰余銘李有志王尚明張志民
中國獸藥雜志 2023年11期
關鍵詞:特征
張傳津,牛華星,韓會剛*,戰余銘 ,李有志,王尚明,張志民
(1.山東省飼料獸藥質量檢驗中心,濟南 250010;2.山東迅達康獸藥有限公司,濟南 250306)
板青敗毒口服液現收載于《獸藥質量標準》2017年版中藥卷,具有清熱解毒,疏風活血的功效,臨床上主要用于雞傳染性法氏囊病等病毒性疾病的輔助治療[1]。現標準通過四個薄層色譜法,分別采用綠原酸對照、甘草酸銨對照、精氨酸對照、芍藥苷對照開展薄層鑒別,供試品前處理繁瑣,同時展開劑采用乙酸丁酯、甲酸、正丁醇、冰醋酸、乙酸乙酯、三氯甲烷等多種有機溶劑,污染大,薄層展開耗時長,而且方中金銀花、蒲公英、白英都含有或多或少的綠原酸;板藍根、大青葉都含有精氨酸,因此本方法專屬性差,重復性也不好,導致結果不易判定。中藥多組分、多靶點的生物特性是其發揮臨床療效的物質基礎,因此如何對中藥復雜多樣的化學成分體系進行質量評價也是重點難點。指紋圖譜、特征圖譜具有信息量大、特征性強、整體性和模糊性等特點,是公認的現代中藥質量評價最為常用的分析方法之一,可宏觀反映中藥化學信息的整體狀況,實現中藥內在質量的有效表征,從整體上提高了中藥質量控制水平[2-3]。基于此,本實驗通過高效液相色譜法,以方中藥物組分為依據,選取色譜中重要的特征信息,采用乙腈-0.4%磷酸溶液系統建立了板青敗毒口服液特征圖譜分析方法,為全面有效控制板青敗毒口服液內在質量提供有力的技術支撐。
1 儀器與材料
1.1 儀器 AE240電子分析天平(梅特勒-托利多儀器有限公司);WatersE2695高效液相色譜儀,二極管陣列PDA檢測器(美國沃特世公司);KQ-400DE數控超聲波清洗器(昆山禾創超聲儀器有限公司);Milli-Q型超純水儀(AdvantageA10)。
1.2 材料 乙腈為色譜純,磷酸為優級純(含量大于85%),甲醇為分析純,水為超純水;對照品信息、樣品信息分別見表1、表2。
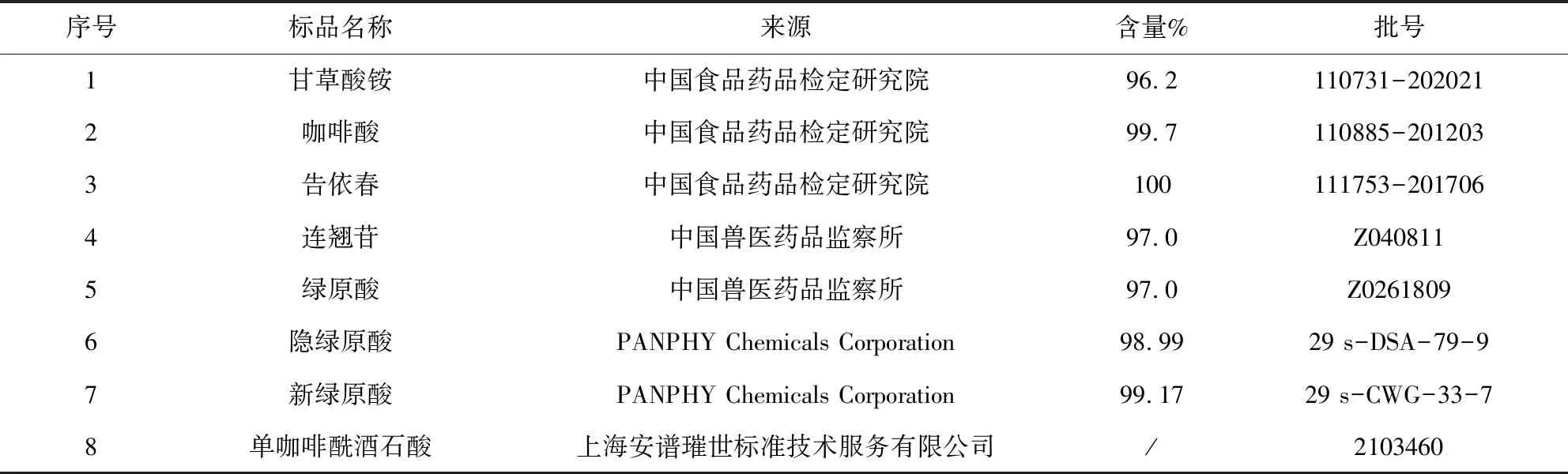
表1 對照品信息表

表2 板青敗毒口服液樣品信息表
2 方法與結果
2.1 色譜條件與系統適用性試驗 采用Waters XBridge C18色譜柱(粒徑為3.5 μm,4.6 mm×250 mm);以乙腈流動相A相,以0.4%磷酸溶液為流動相B,按表3中的規定進行梯度洗脫;檢測波長為240 nm,流速為1.2 mL/min,柱溫為35 ℃,進樣體積10 μL,理論板數按綠原酸峰計算應不低于10000。
2.2 參照物溶液的制備 分別取新綠原酸對照品、綠原酸對照品、隱綠原酸對照品、單咖啡酰酒石酸對照品、(R,S)-告依春對照品、甘草酸銨對照品、連翹苷對照品、咖啡酸對照品適量,精密稱定,置棕色量瓶中,加70%甲醇制成每1 mL各含新綠原酸50 μg、綠原酸50 μg、單咖啡酰酒石酸50 μg、(R,S)―告依春2 μg、咖啡酸10 μg、隱綠原酸50 μg、甘草酸銨50 μg、連翹苷10 μg的混合溶液,即得。
2.3 供試品溶液的制備 精密量取本品2 mL,置50 mL棕色量瓶中,加入50%甲醇適量,超聲處理20分鐘(功率500 W,頻率40k Hz),放冷,加50%甲醇至刻度,搖勻,濾過,取續濾液,即得。
2.4 檢測波長的選擇 以PDA檢測器3D掃描190~450 nm波長范圍,通過不同波長色譜圖的對比發現,在240 nm處各種化合物均有良好的響應值,且各峰分離良好,最終選擇240 nm作為檢測波長。
2.5 方法學考察
2.5.1 精密度試驗 取同一份樣品,連續進樣6次,每次進樣10 μL,按2.1項下色譜條件測定,各色譜峰相對保留時間的RSD< 0.2%,各色譜峰峰面積的RSD<0.4%,結果表明儀器精密度良好。
2.5.2 穩定性試驗 取同一份樣品,按上述色譜條件測定,分別在 0、2、4、6、8、10、12 h進樣10 μL,各色譜峰相對保留時間的RSD<0.12%,結果表明板青敗毒口服液樣品穩定。
2.5.3 重復性試驗 對同一批供試品樣品,重復配制6份供試品溶液,各取10 μl注入色譜儀,將色譜圖數據導入國家藥典委員會“中藥色譜指紋圖譜相似度評價系統(2012版)”,以對照色譜圖為參照圖譜,采用平均數、時間窗為0.6,計算6份樣品相似度,結果顯示其相似度均大于0.999,表明重復性良好,符合要求,結果見表4。
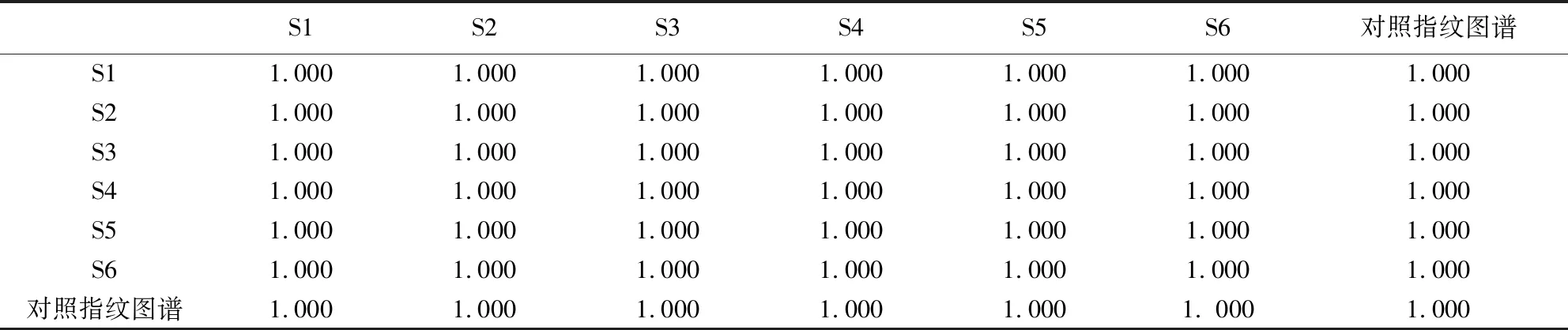
表4 重復性相似度計算結果
2.5.4 不同色譜柱耐用性 分別使用waters XBridge C18(3.5 μm,4.6 mm×250 mm)、賽默飛世爾科技(中國)有限公司Syncronis C18(5 μm,4.6 mm×250 mm)兩根色譜柱,對6批供試品進行測定,結果相似度(S12)均大于0.995,符合特征圖譜技術要求(應不低于0.95)。
2.6 板青敗毒口服液特征圖譜的建立及相似度評價
2.6.1 特征圖譜的建立 按2.1項下色譜條件,測定混合參照物溶液及6批板青敗毒口服液樣品,將色譜圖的數據導入“中藥色譜指紋圖譜相似度評價系統(2012版)”,以S1 為參照圖譜,進行自動匹配,計算6批板青敗毒口服液與對照圖譜之間的相似度。共有8個特征峰,參照物色譜圖及樣品色譜圖見圖1,不同批次樣品疊加特征圖見圖2。
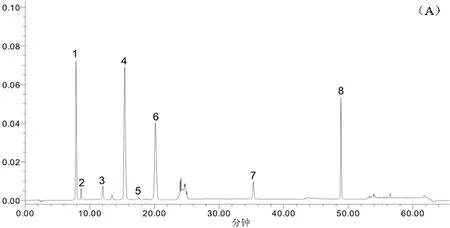
(A.對照品 B.板青敗毒口服液樣品;1.新綠原酸,2.(R,S)告依春,3.單咖啡酰酒石酸,4.綠原酸,5.咖啡酸;6.隱綠原酸;7.連翹苷;8.甘草酸銨)
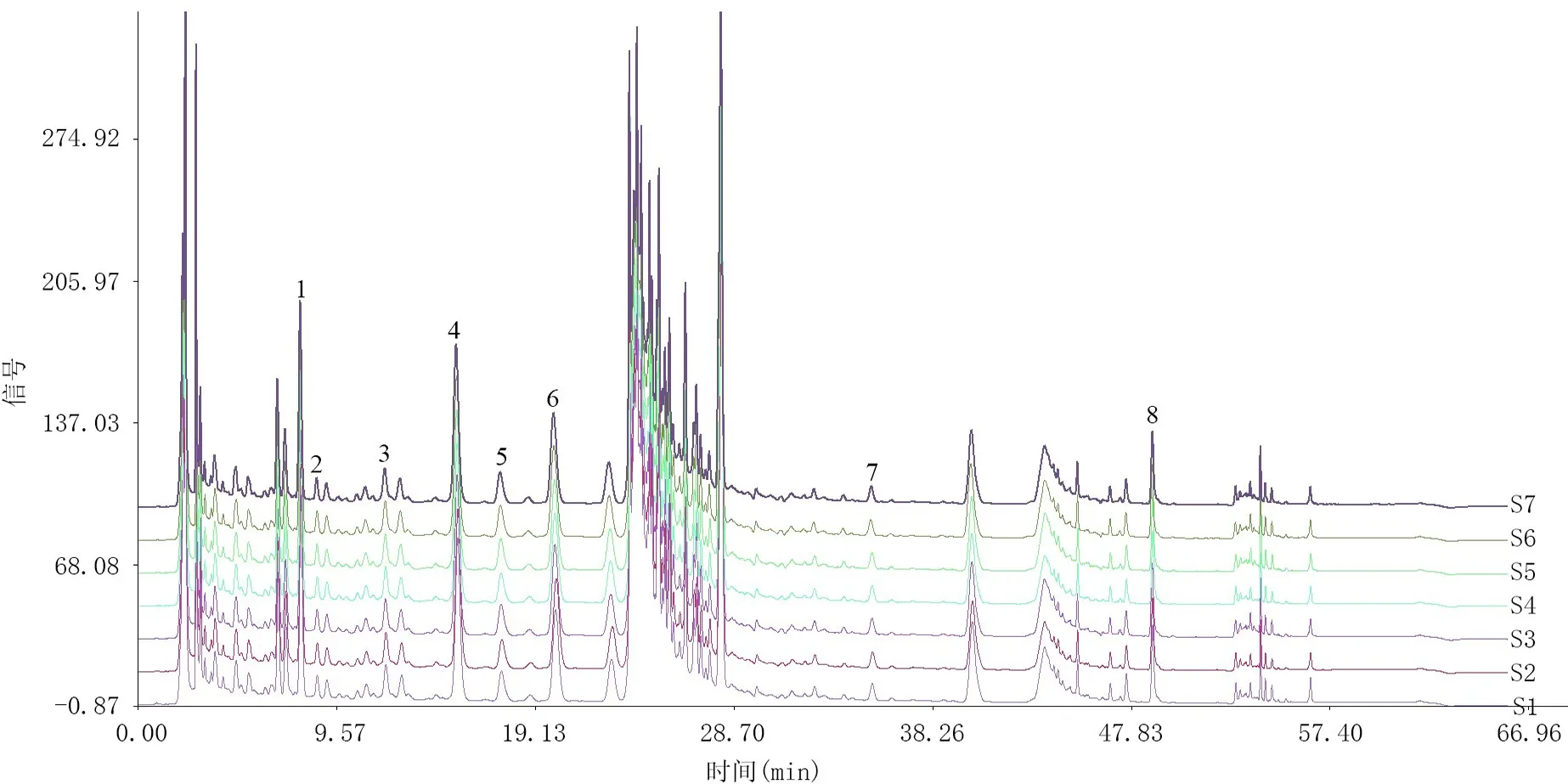
圖2 板青敗毒口服液不同批次疊加圖譜
2.6.2 共有特征峰的確認 將參照物溶液中與板清敗毒口服液中相應的8個共有峰,通過色譜圖與光譜圖比對均有很好的重合性。同時確認1號峰為新綠原酸,2號峰為(R,S)-告依春,3號峰為單咖啡酰酒石酸,4號峰為綠原酸,5號峰為咖啡酸;6號峰為隱綠原酸,7號峰為連翹苷,8號峰為甘草酸銨。板青敗毒口服液處方中金銀花、白英藥材中均有較高含量的綠原酸,蒲公英藥材也含有少量綠原酸,所以綠原酸在口服液中的含量相對較高,出峰時間也較適宜,因此,設定為參考峰S,并計算各特征峰與S峰的相對保留時間。結果顯示,各特征峰的相對保留時間RSD<1.5%,表明具有較好的重現性。按照技術要求,初步得到板清敗毒口服液的特征圖譜的技術參數(表5)。
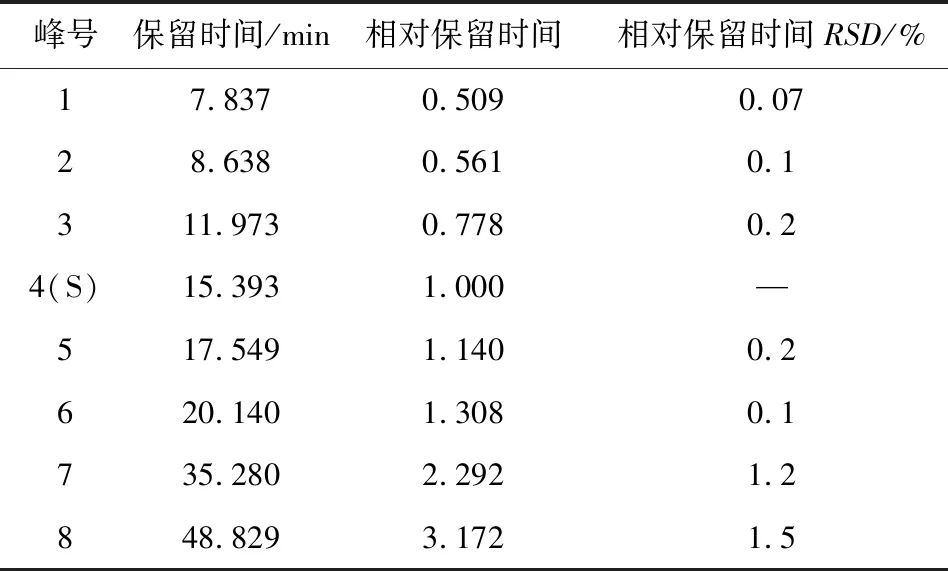
表5 板清敗毒口服液HPLC特征圖譜中特征峰保留時間
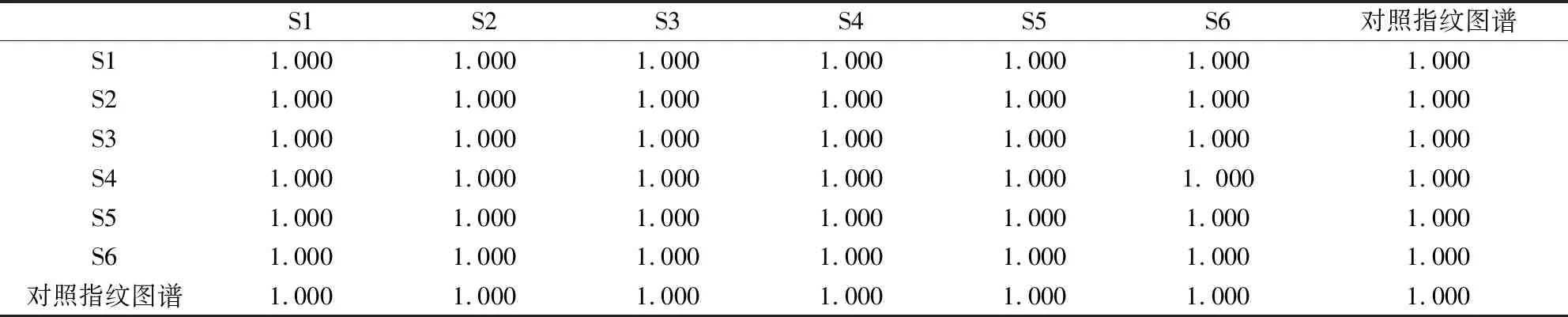
表6 特征圖譜與對照圖譜相似度計算結果
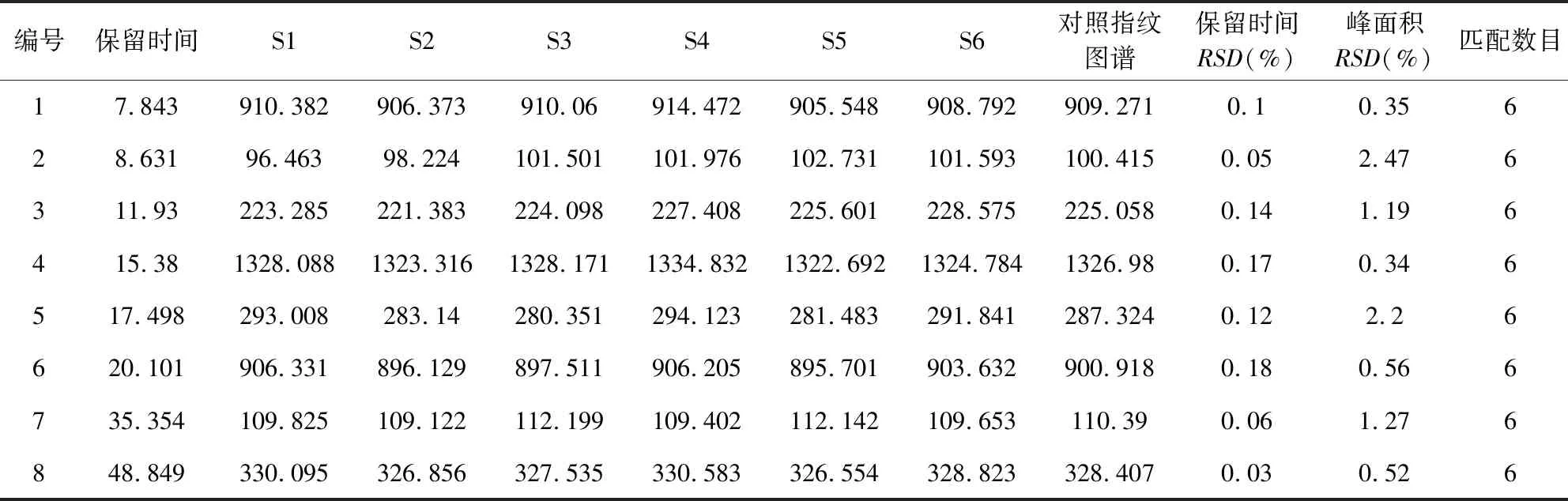
表7 樣品特征圖譜與對照圖譜相似度匹配結果
2.6.3 相似度評價結果 將6批板清敗毒口服液樣品數據導入“中藥色譜指紋圖譜相似度評價系統(2012版)”,得出相似度評價結果表(表4、表5)。
3 討論與結論
3.1 檢測成份的確定 板青敗毒口服液處方包含金銀花、大青葉、板藍根、蒲公英、白英、連翹、甘草、天花粉、白芷、防風、赤芍、浙貝母12味中藥,我們對單味藥材進行處理,分析檢測,結合相關文獻選擇各種藥材合適的質控指標組分。白英(Solanum lyratum Thunb)為茄科植物白英的全草,主要含皂苷類、甾體類化合物、有機酸、黃酮類等組分,其中綠原酸含量較高,也含有少量的咖啡酸[4-6],這兩種組分都是《中國獸藥典》常采納的質控指標組分;金銀花是大宗常用藥材,隨著現代藥理研究的深入,金銀花的質控指標性成分已達15種之多,分別是有機酸酸類(綠原酸、新綠原酸、隱綠原酸、咖啡酸、異綠原酸A、異綠原酸 B、菊苣酸、3,5-二-O-咖啡酰奎寧酸、4,5-二-O-咖啡酰奎寧酸等)、黃酮類(金絲桃苷、蘆丁、木犀草苷、槲皮素等)、環烯醚萜類(馬錢苷酸、馬錢苷、7-表-馬錢苷、獐芽菜苷、斷氧化馬錢苷等)[7-9],其中新綠原酸、綠原酸、隱綠原酸是金銀花含量較高的酚酸類組分,也是《中國藥典》采納的主要質控指標組分[10];蒲公英為菊科多年生草本植物,具有清熱解毒、消腫散結、利尿通淋的功效[11],現代藥理研究表明,蒲公英主要含多酚酸類、萜類、植物甾醇類、倍半萜內酯類和香豆素類等多種化學成分[12],其中酚酸類化合物是蒲公英發揮藥效的重要物質基礎,目前從蒲公英中分離得到的酚酸類化合物有32種,其中單咖啡酰酒石酸、綠原酸、咖啡酸、阿魏酸和菊苣酸是含量較高的5個主要有效成分[13]。《中國藥典》2020年版以菊苣酸的含量作為蒲公英質量評價指標[10],《中國獸藥典》2020 年版以咖啡酸的含量作為蒲公英質量評價指標[14];板藍根、大青葉所含有的氨基酸類、核苷類、黃酮類、嘌呤、靛藍、靛玉紅等成份是近年來質量控制的常用指標成份[15-18],其中(R,S) - 告依春、尿苷、鳥苷、腺苷是板藍根特征圖譜研究優選組分[14,19-20]。綜上所述,本文前期摸索中,擬實現檢測目標:其中板藍根、大青葉:(R,S)-告依春、腺苷;金銀花、蒲公英和白英:新綠原酸、單咖啡酰酒石酸、隱綠原酸、綠原酸、咖啡酸、異綠原酸B、菊苣酸、3,5-二-O-咖啡酰奎寧酸和4,5-二-O-咖啡酰奎寧酸,其中單咖啡酰酒石酸是本方中蒲公英特有的組分;甘草:甘草酸和甘草苷;連翹:連翹苷、連翹酯苷A;赤芍:芍藥苷[14]。后期根據實際樣品檢測情況,基質背景干擾、分離度和各組分含量高低,刪掉了部分目標化合物,成功建立了板青敗毒口服液中8種有效成分(新綠原酸、(R,S)告依春、單咖啡酰酒石酸、綠原酸、咖啡酸、隱綠原酸、連翹苷、甘草酸銨)的特征圖譜。
3.2 檢測波長的選擇 本文采用梯度洗脫的方式,PDA檢測器,3D掃描范圍190~450 nm,2D檢測波長,選擇250 nm(芍藥苷最大吸收波長),240 nm(R,S)―告依春最大吸收波長),277 nm(連翹苷最大吸收波長),322 nm(咖啡酸最大吸收波長),327 nm(綠原酸等系列化合物最大吸收波長),251 nm(甘草酸最大吸收波長),329 nm(單咖啡酰酒石酸、菊苣酸最大吸收波長)328 nm(連翹酯苷A、最大吸收波長)等。由于(R,S)-告依春最大吸收波長240 nm,而在251 nm、324 nm、327 nm、330 nm等處吸收太小,因(R,S)-告依春在方中含量較低,所在優選它的最大吸收波長處,同時我們發現在240 nm處各種化合物均有良好的響應值,且各峰分離良好,最終選擇240 nm作為檢測波長。
3.3 色譜柱的選擇 本文先后測試6根色譜柱,分別為英國ACE EXCEL3 C18-AMIDE(3.0 μm,4.6×150 mm),Agilent Eclipse plus C18(5 μm,4.6×250 mm)、Agilent ZORBAX SB-C18(5 μm,4.6×250 mm)、waters XBridge C18(3.5 μm,4.6×250 mm)、waters AtlantisT3(5 μm,4.6×250 mm)、賽默飛世爾科技(中國)Syncronis C18(5 μm,4.6×250 mm),結果只有XBridge C18、Syncronis C18兩種色譜柱分離度符合要求,進一步進行耐用性實驗,對6批供試品進行測定,結果相似度均大于0.995;而其余色譜柱未達到基線分離,分離度不符合要求。因此本實驗需要指定色譜柱型號,分別為Syncronis C18(5 μm,4.6 mm×250 mm)、XBridge C18(3.5 μm,4.6 mm×250 mm)兩種色譜柱。
板青敗毒口服液由金銀花、大青葉、板藍根、蒲公英、白英、連翹、甘草、天花粉、白芷、防風、赤芍、浙貝母共12味藥組成,經提取精制而成的口服溶液劑。本實驗首次采用HPLC-PDA法,選取方中金銀花、蒲公英、白英、甘草、連翹這些藥材比較重要的8個特征指標成分,建立板青敗毒口服液 HPLC 特征圖譜,作為控制板青敗毒口服液質量的鑒別手段,靈敏度高,重復性好,確保其內在質量的均一、穩定。綜上所述,采用指紋圖譜、特征圖譜能夠反映中藥多成分特點,在中藥質量控制中具有優勢,也是世衛組織認同的中藥質量評價方法,是中獸藥標準提升的有效手段。本方法供試品前處理及流動相僅用到甲醇、乙腈兩種有機溶媒,對環境友好,且前處理簡單,大大縮短檢測時間,提高了工作效率。可作為板青敗毒口服液有效的鑒別和質量評價方法。
猜你喜歡
數學小靈通·3-4年級(2024年2期)2024-05-15 02:02:28
中學生數理化(高中版.高考數學)(2022年3期)2022-04-26 14:04:16
數學年刊A輯(中文版)(2020年1期)2020-05-19 00:30:36
空間科學學報(2020年2期)2020-04-01 03:50:40
瘋狂英語·新策略(2019年10期)2019-12-13 08:43:28
中等數學(2019年8期)2019-11-25 01:38:14
當代陜西(2019年10期)2019-06-03 10:12:04
新聞傳播(2018年11期)2018-08-29 08:15:24
數學小靈通·3-4年級(2017年9期)2017-10-13 08:10:54
廣西科技大學學報(2016年1期)2016-06-22 13:10:38
