氯化膽堿-乙二醇深共熔體系電沉積Ni-Mo 合金鍍層動力學及其電催化析氫性能
2023-12-18 07:24:40盛施展李文暢王慧華
材料工程 2023年12期
盛施展,李 林,李文暢,王慧華
(蘇州大學 沙鋼鋼鐵學院,江蘇 蘇州 215021)
氫能源因熱值高、零污染等優點而成為重要的清潔能源[1-2],其中,電解水制氫因原料來源廣泛、過程清潔環保而備受制氫工業青睞[3-5]。然而,電解水制氫因為電極界面電阻大、能耗高、產氫效率低等缺點,嚴重制約了其快速發展。貴金屬Pt 及其合金因為特殊的能帶結構可加快電解水反應速率,提高產氫效率,但因為儲量有限及價格昂貴,無法大規模應用,因此,開發低成本、高效的析氫催化材料刻不容緩。根據Engel-Brewer 價鍵理論,Ni-Mo 合金對于析氫反應具有良好的電催化協同作用[6-7],成為眾多研究者關注的一個熱點。為進一步提高Ni-Mo 合金電催化析氫性能,研究人員嘗試通過調控Ni-Mo 合金成分或優化微觀形貌提高其本征活性或表觀活性。Zhang 等[8]研究發現,花狀微納米結構的Ni-Mo 合金催化劑具有優異的電催化析氫性能。Zhang 等[9]研究結果表明,Ni-Mo固溶體同樣具有良好的析氫催化活性,10 mA·cm-2的析氫過電位僅為60 mV。深共熔溶劑具有特殊的離子結構,利用其作為溶劑電沉積制備Ni-Mo 合金鍍層有望對合金形貌及成分進行綜合調控,從而實現合金催化性能的大幅提升。Gao 等[10]從氯化膽堿-尿素深共熔體系中電沉積Ni-Mo/Cu 催化材料,其在HER 中的Tafel 斜率為49 mV·dec-1,20 mA·cm-2的過電位僅為63 mV,明顯優于水溶液獲取的Ni-Mo 合金催化性能[8-9]。此外,Florea 等[11]發現,深共熔體系中電沉積Ni-Mo 合金鍍層不僅具有良好的電催化性能,而且與基底具有較好的界面結合力,可以顯著提高合金催化穩定性。目前,有關深共熔體系電沉積Ni-Mo 合金鍍層的報道較少,其沉積動力學也缺乏系統的研究。眾所周知,深共熔溶劑無水或少水,其內部離子結構類型和離子遷移速率與傳統水溶液明顯不同,鐵族金屬去極化或原子氫吸附還原等熟知的經典理論已不再適用于深共熔體系[12-14],因此探討Ni-Mo 合金在深共熔體系中的沉積動力學,尤其合金成分及形貌的演變規律對于制備高性能Ni-Mo 雙金屬合金催化劑具有重要意義。
基于此,本工作以Cu 片為基體,在氯化膽堿-乙二醇(ChCl-EG)深共熔體系中電沉積制備Ni-Mo 合金鍍層,通過交流阻抗譜(EIS)探究Ni-Mo 合金鍍層沉積動力學,借助XRD,EDS 以及SEM 分析沉積產物成分及其形貌演變規律,推測電位驅動下的Ni-Mo 合金反應路徑,并對不同反應路徑產物進行析氫性能表征,旨在理解氯化膽堿-乙二醇深共熔體系中電沉積參數對Ni-Mo 合金鍍層電催化析氫性能的影響規律。
1 實驗材料與方法
1.1 Ni-Mo 合金鍍液配制
實驗原料為氯化膽堿(HOC2H4N(CH3)3Cl,ChCl),乙二醇(C2H6O2,EG),六水氯化鎳(NiCl2·6H2O),一水檸檬酸(C6H8O7·H2O)和四水鉬酸銨(NH4)6Mo7O24·4H2O,所有原料均為分析純,購于阿拉丁試劑有限公司。將氯化膽堿和乙二醇按照摩爾比1∶2,70 ℃均勻混合后形成無色透明的溶劑(30 mL),然后加入1.209 g 的C6H8O7·H2O,充分攪拌至溶液澄清時加入1.112 g (NH4)6Mo7O24·4H2O,繼續攪拌至淡黃色透明溶液,最后加入4.279 g NiCl2·6H2O,攪拌至均勻透明的綠色溶液(Ni-Mo 合金鍍液),待用。
1.2 Ni-Mo 合金鍍層阻抗測試
將除油、酸洗后的潔凈黃銅片作為工作電極,Pt片為對電極,Ag 絲為參比電極,將三根電極垂直置于70 ℃的Ni-Mo 合金鍍液中靜置15 min,期間不斷向電解槽中通入Ar 氣,以減少鍍液中溶解O2對實驗結果的影響,然后進行交流阻抗測試。交流阻抗采用普林斯頓電化學工作站(P3000A),測試頻率范圍為0.001~10000 Hz,測試電位分別為-0.6,-0.8,-1.0,-1.2,-1.4,-1.5 VvsAg。
1.3 Ni-Mo 合金鍍層制備及性能表征
1.3.1 Ni-Mo 合金鍍層制備
采用標準三電極體系電沉積制備Ni-Mo合金鍍層,其中以處理后的黃銅片(1 cm×1 cm,剩余部分用AB 膠密封)為工作電極,Pt片為對電極,Ag絲為參比電極,沉積溫度為70 ℃,沉積時間為5 min,沉積電位參考阻抗測試電位,即-0.6,-0.8,-1.0,-1.2,-1.4,-1.5 V,對應的合金鍍層分別命名為Ni-Mo-0.6,Ni-Mo-0.8,Ni-Mo-1.0,Ni-Mo-1.2,Ni-Mo-1.4 和Ni-Mo-1.5。
1.3.2 Ni-Mo 合金鍍層性能表征
采用SU5000 型場發射電子顯微鏡對鍍層進行微觀形貌分析;借助EDS 能譜儀對鍍層成分進行半定量分析;利用X 射線衍射儀(Ultima Ⅳ)對鍍層表面進行物相分析;采用UV7504 型紫外-可見分光光度計表征合金鍍液的離子類型;利用X 射線光電子能譜(XPS)分析合金鍍層表面元素的化學態,靶材為Mg 靶,發射頻率為1283.3 eV;采用Fischer-MPO 膜厚儀測量鍍層厚度,并計算相應的沉積速率;使用標準三電極體系測試合金鍍層電催化析氫性能,其中Hg/HgO/OH-為參比電極,石墨棒為對電極,Ni-Mo 合金鍍層為工作電極,電解液為1 mol/L KOH,為方便對比,Ni-Mo 合金電催化析氫測試過程中的電位均換算成可逆氫電極,即PRHE=PHg/HgO/OH-+0.098+0.0591pH。
2 結果與討論
2.1 Ni-Mo 合金鍍層的沉積動力學
圖1 為Ni-Mo 合金鍍液的紫外-可見吸收光譜和CV 掃描曲線。由圖1(a)的UV-Vis 譜圖可以看出,Ni-Mo 合金鍍液中有四個特征吸收峰,分別位于λ0=218 nm,λ1=396 nm,λ2=656 nm 和λ3=734 nm 處,對比單一(NH4)6Mo7O24和NiCl2溶液的特征吸收峰位置可知,Ni-Mo 合金鍍液的特征吸收峰僅為(NH4)6Mo7O24和NiCl2溶液吸收峰的疊加,說明合金鍍液中絡合離子類型是由鎳絡合離子和鉬絡合離子機械混合而成。參考文獻[15]可知,200~300 nm 為[Mo7O24(C6H7O7)]5-絡合離子特征吸收峰,350~450 nm 為[Ni(EG)3]2+絡合離子的特征吸收峰,600~700 nm 和700~800 nm 皆為[NiCl4]2-絡合離子的特征吸收峰,進一步說明Ni-Mo 合金鍍液中同時存在[Ni(EG)3]2+,[NiCl4]2-和[Mo7O24(C6H7O7)]5-三種絡合離子。圖1(b)為Ni-Mo 合金鍍液在實驗電位區間內的CV 曲線,可以看出,Ni-Mo 合金鍍液僅表現出一個較寬的還原峰,其起始電位在-0.6 V 處。對比ChCl-EG 溶劑和NiCl2鍍液CV 掃描曲線可知,ChCl-EG 溶劑在-0.2~-1.4 VvsAg 沒有觀察到明顯的還原峰,提高極化電位時,僅表現出歐姆降,說明在實驗電位范圍內ChCl-EG 溶劑是穩定的,而NiCl2鍍液在-0.9~-1.05 V 和-1.05~-1.12 V 處分別出現B1和B2兩個還原峰。結合圖1(a)可知,NiCl2鍍液中僅含有[Ni(EG)3]2+和[NiCl4]2-兩種絡合離子,因此圖中B1和B2分別對應于[Ni(EG)3]2+和[NiCl4]2-的還原[16]。很明顯,Ni-Mo 合金鍍液的還原峰起始電位相比于B1正向移動,說明鍍液中的鉬酸根離子可以促進鎳絡合離子的還原。另外,Ni-Mo 合金鍍液在較寬的電位區間并未觀察到其他明顯的還原峰,說明合金中Mo 與Ni可能發生共沉積行為。
圖2 為Ni-Mo 合金鍍液不同極化電位下的交流阻抗譜圖。從圖2(a),(b)可以看出,在較低的極化電位(-0.6 V 和-0.8 V)時,整個阻抗譜僅表現出被壓扁的容抗弧和典型的擴散弧,并且其變形程度隨極化電位增加越發明顯,說明在此電位區間鍍層呈現多孔態,而且鍍層的沉積速率受鍍液中離子遷移速度控制。-1.0 V 和-1.2 V 極化電位時(圖2(c),(d)),阻抗譜在較低的頻率開始出現感抗弧,如頻率<0.14 Hz(-1.0 V)或頻率<82 mHz(-1.2 V)時均出現明顯的感抗弧,且其特征頻率隨極化電位增加而增大(10 mHz 增大至26 mHz),說明電極表面開始有中間吸附體存在,且中間吸附體數量隨極化電位增加逐漸增多。進一步提高極化電位(-1.4 V 和-1.5 V)(圖2(e),(f)),可觀察到感抗弧減小甚至消失,說明電極表面中間吸附體數量減少或完全被消耗。通常,中間吸附體反應速率受離子擴散速度以及電荷轉移速度共同控制,極化電位低,離子擴散速度大于電荷轉移速度,中間吸附體可在電極表面積累,因此表現出明顯的感抗弧;極化電位高,電荷轉移速度大于離子擴散速度,中間吸附體反應消耗快,電極表面積累量減少,感抗弧減小或消失。此外,在高極化電位時較低頻率段重新出現新的容抗弧,這可歸結為合金鍍液中游離出來的H+被還原產生氫氣的過程[13]。
圖3 為Ni-Mo 合金鍍層在不同極化電位下(-0.8,-1.0,-1.2,-1.4 V 和-1.5 V)的SEM 圖和EDS分析。可以看出,Ni-Mo 合金鍍層的微觀形貌受極化電位影響較大,極化電位為-0.8 V 時(圖3(a-1),(a-2)),鍍層表面較為平整,伴有針刺邊緣的粒狀顆粒呈現松散堆積,該電位下的合金鍍層主要由金屬Ni 組成(圖3(a-3)),少量的O 可歸因于鍍層表面吸附氧或金屬Ni 氧化所致。當極化電位增加到-1.0 V 時(圖3(b-1),(b-2)),鍍層表面依舊平整,但顆粒呈現明顯的針狀結構,說明增加極化電位有利于晶體擇優生長,此時合金仍然以金屬Ni 為主(圖3(b-3))。當極化電位進一步增加至-1.2,-1.4 V 和-1.5 V 時(圖3(c-1),(c-2),(d-1),(d-2),(e-1),(e-2)),Ni-Mo 合金鍍層表面形貌發生顯著變化,呈現出由多個細小晶粒聚集而成的胞狀晶,且極化電位越大,胞狀晶的晶粒尺寸越大,鍍層表面的裂紋越發明顯,合金中Mo 含量越高(圖3(c-3),(d-3),(e-3)),說明Ni-Mo 合金中的Mo 含量是引起表面形貌改變和鍍層內應力提升的重要原因。此外,合金中的O 含量隨著極化電位增加先上升后下降,在-1.2 V 時達到最大,結合圖2(d)可知,在-1.2 V 時電極表面有大量中間吸附體富集,推斷在該電位下[Mo7O24(C6H7O7)]5-開始被還原,生成MoOx等中間吸附體,但隨著極化電位增加,中間吸附體還原速度提高,合金中的O 含量又呈現下降趨勢(圖3(e-3))。

圖3 Ni-Mo 合金鍍層SEM 照片和EDS 分析(a)Ni-Mo-0.8;(b)Ni-Mo-1.0;(c)Ni-Mo-1.2;(d)Ni-Mo-1.4;(e)Ni-Mo-1.5;(1)低倍;(2)高倍;(3)EDS 分析Fig.3 SEM images and EDS analysis of Ni-Mo alloy coatings(a)Ni-Mo-0.8;(b)Ni-Mo-1.0;(c)Ni-Mo-1.2;(d)Ni-Mo-1.4;(e)Ni-Mo-1.5;(1)low magnification;(2)high magnification;(3)EDS analysis
圖4 為不同極化電位下Ni-Mo 合金鍍層的XRD 分析。可以看出,在低極化電位時(-0.8 V和-1.0 V),合金鍍層表面呈現(111)Ni和(200)Ni衍射峰,且極化電位越大,Ni 衍射峰強度越高,表明結晶形態越好;隨著極化電位的增加(-1.2 V),(111)Ni峰形明顯寬化,且向低角度微弱偏移,說明有少量的Mo 固溶到Ni 結構中引起晶格膨脹(Mo≈18.6%,質量分數,下同);隨著極化電位進一步增加(-1.4 V 和-1.5 V),(111)Ni衍射峰消失,左側附近出現較小的饅頭峰,說明形成具有非晶形態的Ni-Mo 合金。結合圖3(e-3)對應的EDS能譜分析結果,可以估算出合金中Ni/Mo 化學計量比約為4,因此Ni-Mo 合金鍍層在較高極化電位下可形成Ni4Mo 非晶合金。

圖4 不同極化電位下Ni-Mo 合金鍍層的XRD 分析Fig.4 XRD analysis of Ni-Mo alloy coatings fabricated at different polarization potentials
圖5 進一步給出Ni-Mo-1.4 合金鍍層表面元素化學態分析。從圖5(a)可以看出,Ni2p 高分辨圖譜可以擬合成五個峰,其中851.98 eV 對應金屬態Ni0的特征峰,而位于855.94 eV 和873.51 eV 的兩個擬合峰分別對應于Ni—O 的2p3/2和2p1/2自旋軌道特征峰,并伴有典型的衛星峰。Mo3d 圖譜中有三個擬合峰(圖5(b)),其中結合能為227.7 eV 對應于金屬態Mo0特征峰,232.34 eV 和235.49 eV 分別對應于不同價態的Mo—O 鍵,該結論進一步說明鍍層除了Ni-Mo 合金外,還夾雜有NiO 和MoOx,這些氧化物可能歸因于活潑金屬的表面氧化,該分析結果與EDS 分析結果基本保持一致。
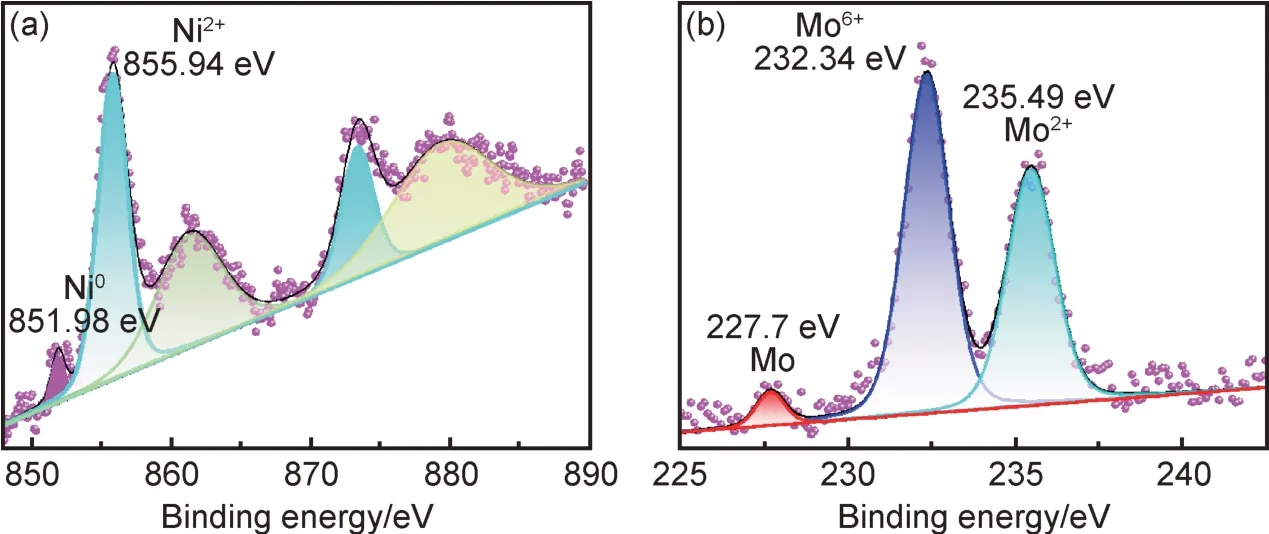
圖5 Ni-Mo-1.4 合金鍍層表面XPS 高分辨譜圖 (a)Ni2p;(b)Mo3dFig.5 XPS high-resolution spectra of Ni-Mo-1.4 alloy coatings (a)Ni2p;(b)Mo3d
綜上,初步判定Ni-Mo 合金鍍層的沉積動力學為:在低的極化電位時(≤-1.0 V),合金鍍液主導鎳絡合離子的還原,鉬酸鹽中間價態產物(MoOx)形成相對困難,電極表明呈現多孔的納米鎳鍍層,表現為阻抗譜高頻段壓扁的容抗弧(圖2(a),(b)),電極反應式如式(1)和式(2);當極化電位進一步增加時,電極附近的[Mo7O24(C6H7O7)]5-絡合離子開始發生還原反應,生成中間價態的MoOx,造成合金鍍層中的Mo 和O 含量顯著增加(圖3(c-3))。根據文獻[11-12],鉬酸鹽還原的中間產物主要為MoO2,且電極附近的Ni2+將進一步促進MoO2轉化成鎳鉬氧化物(MoO2Ni4),電化學反應式見式(3)和式(4)。此外,合金鍍液中檸檬酸因配位數發生變化而離解出的H+在該極化區間容易被還原成H 原子(反應式(5)),并聚集形成H2通過電極表面的中間吸附體間隙不斷逸出,但隨著極化電位進一步增加,中間吸附體厚度明顯增加,H2逸出能力下降,大量氫原子滯留在中間吸附體內部,因此在H 原子作用下,中間吸附體會被還原成Ni-Mo合金,而根據圖3(d-3),(e-3)能譜分析可知,最終合金鍍層中Ni/Mo 化學計量比約為4,因此推斷電極表面發生的反應式如式(6)所示。需要指出,盡管在高極化電位下Ni-Mo 合金鍍層接近Ni4Mo 組成,但是鍍層的晶化程度較低,仍然以非晶形態存在(圖4)。
通常,沉積過程中鍍層的內應力(拉應力)常因鍍層厚度增加而增大,而內應力不僅影響鍍層界面結合力,同時誘導鍍層內部出現微裂紋,從而影響鍍層催化穩定性和催化活性。在電鍍過程中,鍍層厚度與沉積速率成正比,而沉積速率受界面電荷轉移速度和鍍液中離子擴散速度共同控制,不同極化電位下電極上電荷轉移速度和溶液中離子擴散速度不一致,造成界面電化學反應速率不同,因而沉積速率也不同。圖6為Ni-Mo 合金鍍層在不同極化電位下的沉積速率。可以看出,鍍層的沉積速率隨著極化電位增加而增大,說明極化電位增大,電極表面的電荷轉移加快,沉積速率增加,但當極化電位增加至一定值后(>-1.4 V),沉積速率減緩,說明此時鍍液中離子擴散速度是速度控制步驟,即鍍液中離子擴散速度決定鍍層的沉積速率。另外,由于在大的極化電位下,鍍層沉積速率較大,厚度顯著增加會導致拉應力快速上升,造成鍍層產生裂紋,如圖3 所示。

圖6 Ni-Mo 合金鍍層在不同極化電位下的沉積速率Fig.6 Deposition rate of Ni-Mo alloy coatings at different polarization potentials
2.2 Ni-Mo 合金鍍層的析氫催化性能
不同極化電位制備的Ni-Mo 合金鍍層不僅成分存在差異,其微觀形貌也明顯不同,兩者均影響Ni-Mo 合金鍍層電催化析氫性能。圖7 為Ni-Mo 合金鍍層析氫催化性能。從圖7(a)的 LSV(linear sweep voltammetry)曲線可知,Ni-Mo 合金鍍層析氫過電位隨極化電位增加先減小后增加,即Ni-Mo-1.4合金鍍層過電位最小,繼續增加極化電位,其析氫過電位又顯著增加。Tafel 斜率是衡量電極析氫動力學的重要參數之一,可根據Tafel 斜率大小判斷析氫反應速度控制步驟,如Volmer步驟主要以電極吸附氫為速度控制步驟(H2O+e+*→H*+OH-,120 mV·dec-1),Heyrovsky 步驟主要以電化學脫附為速度控制步驟(H*+H2O+e →H2+OH-+*,40 mV·dec-1),Tafel步驟主要以化學脫附為速度控制步驟(2H*→H2+*,30 mV·dec-1)[17]。圖7(b)為Ni-Mo 合金鍍層的Tafel斜率,可以看出,Ni-Mo-1.4 合金鍍層具有最小的Tafel 斜率(48.7 mV·dec-1),說明合金鍍層上的析氫反應速度主要受Heyrovsky-Heyrovsky 混合控制。圖7(c)給出了Ni-Mo 合金鍍層在10 mA·cm-2和100 mA·cm-2電流密度下的析氫過電位,可明顯看出Ni-Mo-1.4 合金鍍層的析氫過電位較小(η10=51 mV,η100=149 mV),說明其具有良好的電極反應動力學。圖7(d)為Ni-Mo-1.4 合金鍍層電催化析氫穩定性測試,可以看出,合金循環析氫1000 周次后其電位下降幅度較小(Δη100=11 mV),而且經持續催化后其表面形貌沒有發生明顯變化(圖7(d)插圖),僅微裂紋數量較初始狀態有所增加,說明持續的H2逸出會加劇鍍層內部微裂紋的擴展,同時引起表面少許顆粒剝落。由于鍍層屬于層狀堆積結構,表面微小脫落不會造成Ni-Mo 合金鍍層催化性能的大幅下降,因此利用深共熔體系電沉積制備的Ni-Mo 合金鍍層不僅具有良好的催化活性,同時有著較好的催化穩定性,這些都得益于深共熔溶劑對合金成分的有效調控,并極大地提高鍍層的界面結合力。表1 總結了已有報道的Ni-Mo雙金屬或Ni-Mo-X三元合金析氫催化劑的過電位及Tafel 斜率,通過數據分析,可知氯化膽堿-乙二醇深共熔體系中電沉積制備Ni-Mo/Cu 合金鍍層具有更加優異的電催化析氫性能[18-25]。

表1 Ni-Mo/Cu 與其他鎳基催化劑電催化析氫中的過電位及Tafel 斜率Table 1 Overpotentials and Tafel slopes between Ni-Mo/Cu and other reported Ni-based catalysts for HER

圖7 Ni-Mo 合金鍍層的析氫催化性能(a)LSV;(b)Tafel 斜率;(c)過電位;(d)Ni-Mo-1.4 合金鍍層析氫穩定性Fig.7 HER catalytic performance of Ni-Mo alloy coatings(a)LSV;(b)Tafel slope;(c)overpotential;(d)catalytic stability of Ni-Mo-1.4 alloy coatings
電化學活性面積(electrochemical active surface area,ECSA)也是評價催化劑性能好壞的重要參數之一,通常與電極/溶液界面的雙電層電容(Cdl)成正比[26],即Cdl越大,ECSA 越大,催化劑活性位點越多,催化性能越好。圖8 為Ni-Mo 合金鍍層不同掃描速率下循環伏安曲線及電流密度-掃描速率線性擬合圖。可以看出,掃描速率越大,Ni-Mo 合金鍍層界面雙電層電容充放電電流越大;相同掃描速率下,極化電位越大,雙電層電容充放電電流也越大(圖8(a)~(e)),但極化電位增大至一定值后,電極界面雙電層電容充放電電流開始減小(圖8(f))。圖8(g)給出了不同極化電位下Ni-Mo 合金鍍層界面的雙電層電容,可以看出,Ni-Mo-1.4 合金鍍層的電容最大(Cdl=22.1 mF·cm-2),說明Ni-Mo-1.4 合金鍍層的電化學活性面積最大,在此合金表面可以產生更多的活性位點,因此表現出優異的析氫催化活性。

圖8 Ni-Mo 合金鍍層的電化學活性表征 (a)~(f)Ni-Mo-0.6,Ni-Mo-0.8,Ni-Mo-1.0,Ni-Mo-1.2,Ni-Mo-1.4 和Ni-Mo-1.5 合金鍍層不同掃描速率CV 曲線;(g)雙電層電容;(h)本征交換電流密度Fig.8 Characterization of electrochemical activity for Ni-Mo alloy coatings (a)-(f)CV curves at different scan rates of Ni-Mo-0.6,Ni-Mo-0.8,Ni-Mo-1.0,Ni-Mo-1.2,Ni-Mo-1.4 and Ni-Mo-1.5 alloy coatings;(g)double layer capacitance;(h)intrinsic exchange current density
同理,本征交換電流密度(j0,real)也是衡量電催化劑本征催化活性的一個關鍵指標,而成分是影響其本征活性的重要因素。為進一步理解極化電位對Ni-Mo 合金鍍層本征活性的影響規律,對其本征交換電流進行考察,如圖8(h)所示。本征交換電流密度(j0,real)是表觀交換電流密度(j0,app)與實際工作電極面積(Sreal)的比值(j0,real=j0,app/Sreal),其中Sreal=Cdl/k(k為單位面積金屬汞的雙電層電容,通常為20 μF·cm-2),j0,app=10-a/b(a為Tafel 擬合線的截距,b為Tafel 斜率)[27]。可以看出,Ni-Mo 合金鍍層的本征活性與合金鍍層中Mo 含量成正比,Mo 含量越高,其本征活性越高(Mo=28.6%,j0,real=1.07×10-3mA·cm-2)。值得說明,雖然Ni-Mo-1.5 合金鍍層本征活性較高,但其Tafel 斜率以及過電位較大(圖7(b),(c)),可能因為其組成顆粒粗大,表面粗糙度明顯降低,能夠暴露在外的電化學活性面積較小(Cdl=5.21 mF·cm-2),表觀活性相對較小,綜合催化性能下降。
3 結論
(1)隨著極化電位增加,Ni-Mo 合金鍍層的成分經歷Ni →Ni+MoO2(MoO2Ni4)→Ni4Mo 演變。
(2)隨著極化電位增加,Ni-Mo 合金鍍層析氫過電位先減小后增大,其中Ni-Mo-1.4 合金鍍層析氫過電位最小(η10=51 mV),且具有較小的Tafel 斜率(48.7 mV·dec-1)和較大的電化學活性面積(Cdl=22.1 mF·cm-2),表明其具有良好的析氫催化活性。
(3)Ni-Mo-1.4 合金鍍層表現出良好的催化穩定性,循環析氫1000 周次后電位下降較小(Δη100=11mV),說明深共熔溶劑有助于提升鍍層與基底的界面結合力。
