高效液相色譜-串聯質譜法同時測定地表水中33 種藥物
2023-12-26 10:06:30李慶山卜慶偉曹紅梅吳曉澤郭亞麗姜巍巍
中國環境監測 2023年5期
李慶山,卜慶偉,曹紅梅,洪 宸,吳曉澤,郭亞麗,姜巍巍
1.中國礦業大學(北京)化學與環境工程學院,北京 100083
2.上海城市水資源開發利用國家工程中心有限公司,上海 200082
藥物在疾病預防和治療、生長發育調節以及代謝功能改善等方面都發揮了巨大的作用,但殘留藥物的排放也對人體健康和生態環境產生了潛在的危害[1-3]。 據統計,2013 年我國抗生素總使用量約為16 萬t[4]。 進入人體或動物體內的藥物有50%以上不能被吸收,主要以原藥或代謝產物的形式隨尿液和糞便排出體外[5],導致藥物在地表水、地下水、飲用水、土壤、沉積物、湖泊、海洋等各種環境介質中被廣泛檢出[6-8]。 雖然藥物在地表水中以ng/L ~μg/L 的痕量水平存在,但藥物活性成分在環境中難降解,且能夠誘導耐藥菌群或抗性基因的產生,長期暴露于含有藥物的水環境中還會導致藥物在生物體內累積,對水生生物產生繁殖毒性、生長毒性和免疫毒性等危害[9-10]。為了獲取藥物在水環境中的存在水平、環境歸趨和生態風險等基礎數據,有必要建立環境中各種藥物的分析方法。
目前國內外建立的藥物分析方法主要基于固相萃取-高效液相色譜-串聯質譜法(SPE-HPLCMS/MS),該方法可有效富集、凈化水質樣品,并以高靈敏度檢出而被廣泛應用[11-12]。 比如,王婭南等[13]基于SPE-HPLC-MS/MS 建立了地表水中40 種抗生素的分析方法,采用正、負離子模式分別采集,方法檢出限為0.002 ~0.270 ng/L,測定地表水加標回收率為61.0%~149%,相對標準偏差為1.2%~32%。 FENG 等[14]建立了表層水中5類40 種抗生素的分析方法, 加標回收率為41.3%~112.6%,利用該方法分析長江南京段表層水,共檢測到13 種抗生素,質量濃度為13.4 ~780.5 ng/L。 ZHOU 等[12]建立了11 類50 種抗生素在地表水、湖水、污水、沉積物、糞便和污泥中的分析方法,方法定量限分別為0.52 ~5.88 ng/L、2.36~65.8 ng/L、1.73 ~20 ng/L、0.64 ~6.67 ng/g、1.33~17.4 ng/g、1.50 ~28.6 ng/g,方法回收率為50%~150%。 MAIA 等[15]建立了7 種喹諾酮類抗生素的分析方法,標準曲線線性范圍為50 ~1 300 ng/L,方法檢出限和定量限分別為6.7 ~59.0 ng/L和23.3~196.6 ng/L,該方法被應用于污水中抗生素存在水平的分析。 上述分析方法主要涵蓋磺胺類、四環素類、喹諾酮類、大環內酯類、β-內酰胺類等抗生素類藥物,但是除抗生素外受納環境還會受其他多種藥物的影響,比如抗病毒藥、消炎藥和調節血脂藥等[16-17]。 特別是近年來受新冠疫情的影響,疫情發生區水環境中抗病毒藥物的檢出率明顯升高[18]。 此外,不同種類藥物的物理化學屬性差異較大,為了提高分析方法的準確性,需要重點對前處理條件進行優化,比如固相萃取柱的選擇、樣品pH、洗脫溶劑類型及用量等[19]。
本研究選擇產量大、環境中檢出率高、毒性強的化合物及抗新冠藥物,確定了25 種抗生素藥物(包括7 種磺胺類、5 種大環內酯類、4 種四環素類、5 種喹諾酮類、1 種β-內酰胺類、3 種其他類)和8 種非抗生素藥物(3 種調節血脂藥、2 種抗病毒藥、1 種抗痙攣藥、1 種消炎藥、1 種抗寄生蟲藥)共33 種目標物。 采用SPE-HPLC-MS/MS 技術,通過優化色譜、質譜條件及前處理方法,建立了測定地表水中33 種藥物的分析方法,并將建立的分析方法應用于北京市涼水河地表水中藥物的分析測定。
1 研究方法
1.1 儀器與試劑
儀器:高效液相色譜-三重四級桿串聯質譜儀(Shimadzu LC/MS-8040,LabSolution 色譜工作站,日本);12 孔固相萃取裝置(Supelco 公司,美國);旋渦混合器(江蘇海門其林貝爾儀器制造有限公司,中國);超聲波清洗儀(江蘇昆山舒美超聲儀器有限公司,中國);氮吹儀(北京帥恩科技有限責任公司, 中國); Oasis HLB 型固相萃取柱(6 mL/500 mg, Waters 公司, 美國);0.22 μm PTFE 膜針式過濾器(Millipore,美國);0.45 μm 玻璃纖維濾膜(直徑142 mm,Millipore,美國);Shimpack XR-ODS 反相色譜柱(2 mm×75 mm,2.2 μm,島津公司,日本),pH 計(Mettler Toledo,瑞士)。
試劑:甲醇、乙腈(色譜純,Fisher,美國);甲酸、氨水、氫氧化鈉(分析純,Aladdin,上海);乙酸銨(分析純,麥克林,上海)。
標準品:磺胺嘧啶(SD)、磺胺二甲嘧啶(SMT)、磺胺甲嘧啶(SMR)、磺胺甲惡唑(SMX)、磺胺間甲氧嘧啶(SMM)、磺胺吡啶(SPD)、甲氧芐啶(TP)、阿奇霉素(ATM)、紅霉素(ETM)、林可霉素(LIN)、 羅紅霉素(RTM)、 克拉霉素(CTM)、金霉素(CTC)、多西環素(DC)、四環素(TC)、土霉素(OTC)、環丙沙星(CPX)、恩氟沙星(EFX)、諾氟沙星(NFX)、氧氟沙星(OFX)、培氟沙星(PFX)、氨芐西林(AMP)、氯霉素(CP)、氟苯尼考(FF)、灰黃霉素(GSV)、貝螺殺(NIC)、利托那韋(RTV)、磷酸氯喹(PCQ)、苯扎貝特(BF)、卡馬西平(CBZ)、氯貝酸(CA)、雙氯芬酸(DF)、吉非羅齊(GF)、磺胺二甲嘧啶-D4(SMT-D4)、卡馬西平-D10 (CBZ-D10)、 羅紅霉素-D7 (RTMD7)、氯霉素-D5(CP-D5)、諾氟沙星-D5(NEXD5)、去甲基金霉素(DMC)。 DC、ETM、GF、SMM購自日本Tokyo Chemical Industry 公司;CTC 購自德國Dr.Ehrenstorfer 公司;LIN 購自英國Apollo Scientific 公司;SMR 購自美國Alfa Aesar 公司;PCQ 購自中國食品藥品檢定研究院;CP-D5、SMTD4、NFX-D5 購自壇墨質檢標準物質中心;RTMD7、 CBZ-D10 購自加拿大 Toronto Research Chemicals 公司;其余標準品均購自北京百靈威科技有限公司。 標準品純度均高于98%,滿足定量分析要求。
標準溶液的配制包括標準儲備液的配制、混合標準儲備液的配制以及混合標準工作液的配制。 標準儲備液的配制:準確稱取10 mg 標準品于10 m L 色譜標樣存儲瓶中,喹諾酮類的藥物(CPX、EFX、NFX、OFX、PFX)先用0.5 m L NaOH(0.1 mol/L)溶解,再用甲醇定容至10 m L。 PCQ直接用去離子水配制,4 ℃下冷藏一周。 其余標準品均用甲醇溶解,配制得到1 mg/m L 標準儲備液,-20 ℃冷凍保存,保存時間為3 個月。 混合標準儲備液的配制:分別移取0.1 m L 標準儲備液于10 m L 色譜標樣瓶, 用甲醇定容, 配制得到10 μg/m L 的混合標準儲備液,-20 ℃冷凍保存,保存時間為一個月。 混合標準工作液的配制:將混合標準儲備液稀釋至適當濃度的混合標準工作液,使用前配制,現配現用。
1.2 分析條件
1.2.1 色譜條件
Shim-pack XR-ODS 反相色譜柱(2 mm ×75 mm,2.2 μm,島津公司,日本);進樣量10 μL;流速0.3 m L/min;柱溫30 ℃;流動相A:0.2%甲酸-2 mmol/L 乙酸銨-水溶液,流動相B:乙腈。 流動相梯度:0 ~5 min,10% ~15% B;5 ~7 min,15%~20% B;7 ~11 m in,20% ~40% B;11 ~14 min,40%~60% B;14 ~16 min,60%~95% B,保持2 m in;18 ~18.1 m in,95%~10% B;18.1 ~22 m in,10% B。
1.2.2 質譜條件
采用 ESI +/ESI-切換, 多反應監測模式(MRM),正離子模式下離子源接口電壓-3.5 kV,負離子模式下離子源接口電壓4.5 kV,溶劑管溫度250 ℃,加熱模塊溫度400 ℃,霧化氣氮氣,流速3 L/min,干燥氣為氮氣,流速15 L/min,碰撞氣為氬氣。
1.3 樣品前處理方法
地表水樣采集運回實驗室后經0.45 μm 玻璃纖維濾膜過濾。 準確量取2 份2 000 m L 子樣品,分別用甲酸、氨水調節pH 至3.0 和9.0,隨后加入100 ng 定量內標(1 μg/mL),充分混勻,采用HLB 固相萃取小柱對水樣中的目標物進行富集。固相萃取柱活化條件依次為8 m L 甲醇、8 m L 高純水,活化完成后,以大約3 ~5 m L/min 的流速將水樣通過固相萃取柱。 富集完成后,用8 m L 的高純水淋洗HLB 柱,抽真空干燥60 m in 以去除殘余水分。 富集pH=3 水樣的HLB 柱用8 m L 甲醇洗脫,富集pH=9 的水樣先用4 m L 甲醇洗脫,再用4 m L 體積分數5%氨化甲醇進行洗脫。 洗脫液分別收集于K-D 濃縮器中,用柔和高純氮氣吹至近干,用初始流動相定容至1 m L,渦旋混合后經PTFE 膜針式過濾器過濾,置于4 ℃冰箱內避光保存,待HPLC-MS/MS 分析。
2 結果與討論
2.1 色譜條件的優化
色譜柱的選擇應首先保證目標物的分離度和分析時間,其次是柱容量。 柱長影響目標物的分離度和分析時間,柱長越長,分離度越高,但分析時間也會延長。 內徑越小意味著柱效越高,分離效果越好,但對檢測器的靈敏度要求越高[20-21]。由于本方法中目標物涵蓋了四環素類、喹諾酮類、大環內酯類、磺胺類等抗生素,每一類又包含了數種性質相似的藥物,為了保證目標物的有效分離,盡量節省分析時間,我們綜合對比了文獻中報道的50 mm×2 mm、75 mm×2 mm、75 mm×3 mm 和100 mm×2 mm 4 種常用的Shim-pack XR-ODS 反相色譜柱[22-23],發現75 mm×2 mm 可滿足本方法的分析要求。 在色譜柱粒徑的選擇上,小粒徑液相色譜柱填料具有柱效高、穩定性強等優點,適用于多種抗生素的分析。 研究發現,在高效液相色譜系統中2.2 μm 顆粒填料比5.0 μm 顆粒填料的柱效高3 倍[23]。 綜合分析,最終選擇Shimpack XR-ODS 色譜柱(2 mm×75 mm,2.2 μm)進行后續實驗。
流動相的選擇以及梯度程序的優化是色譜條件優化的重要環節[13]。 單一的流動相組分無法滿足多種藥物的分離效果,響應強度欠佳。 甲醇或乙腈是目前應用最為廣泛的有機相組成[24],本研究對比了甲醇和乙腈對目標物的分離效果,發現乙腈的洗脫能力更強,對33 種目標物的分離效果較好,故選擇乙腈作為有機相。 以純水作為無機相時,發現喹諾酮類藥物、四環素類藥物及PCQ 有嚴重的拖尾現象,此外,還發現DF、DC、CBZ、GF 等藥物離子化程度較差導致的響應強度低。 加入甲酸可以促進目標物產生[M+H]+分子離子峰,因此考慮向水相中加入甲酸[13],結果發現加入體積分數0.2%甲酸能有效提升正離子模式下目標物的響應強度和靈敏度。 為了使無機相保持弱酸弱堿鹽緩沖體系,并且保證負離子模式下目標物分析的穩定性和準確性,最后向無機相中加入2 mmol/L 乙酸銨。 最終確定流動相的組成:0.2%甲酸-2 mmol/L 乙酸銨-水溶液(流動相A)和乙腈(流動相B)。 18 min 內可將33 種目標物實現有效分離,總離子流色譜圖見圖1。
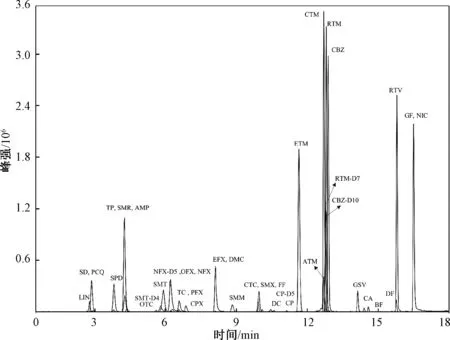
圖1 各目標物及內標物的總離子流色譜圖Fig.1 Total ion chromatogram of the target compounds and internal standards
2.2 質譜條件的優化
將目標物和內標物配制成100 ng/m L 標準溶液,在ESI+/ESI-模式下進行全掃,采用一級質譜進行母離子全掃,選擇峰強度高、穩定的分子離子作為母離子,依次優化碎裂電壓、子離子、碰撞電壓。 SD、SMT、SMR、SMX、SMM、SPD、TP、CBZ、ATM、ETM、 LIN、 RTM、 CTM、 AMP、 GSV、 RTV、PCQ、CTC、DC、OTC、TC、CPX、EFX、NFX、OFX、PFX、SMT-D4、CBZ-D10、RTM-D7、NFX-D5、DMC在正離子模式下響應強度較高。 BF、CA、DF、GF、CP、FF、NIC、CP-D5 在負離子模式下響應強度較高。 33 種目標物的質譜參數、保留時間及內標如表1 所示。
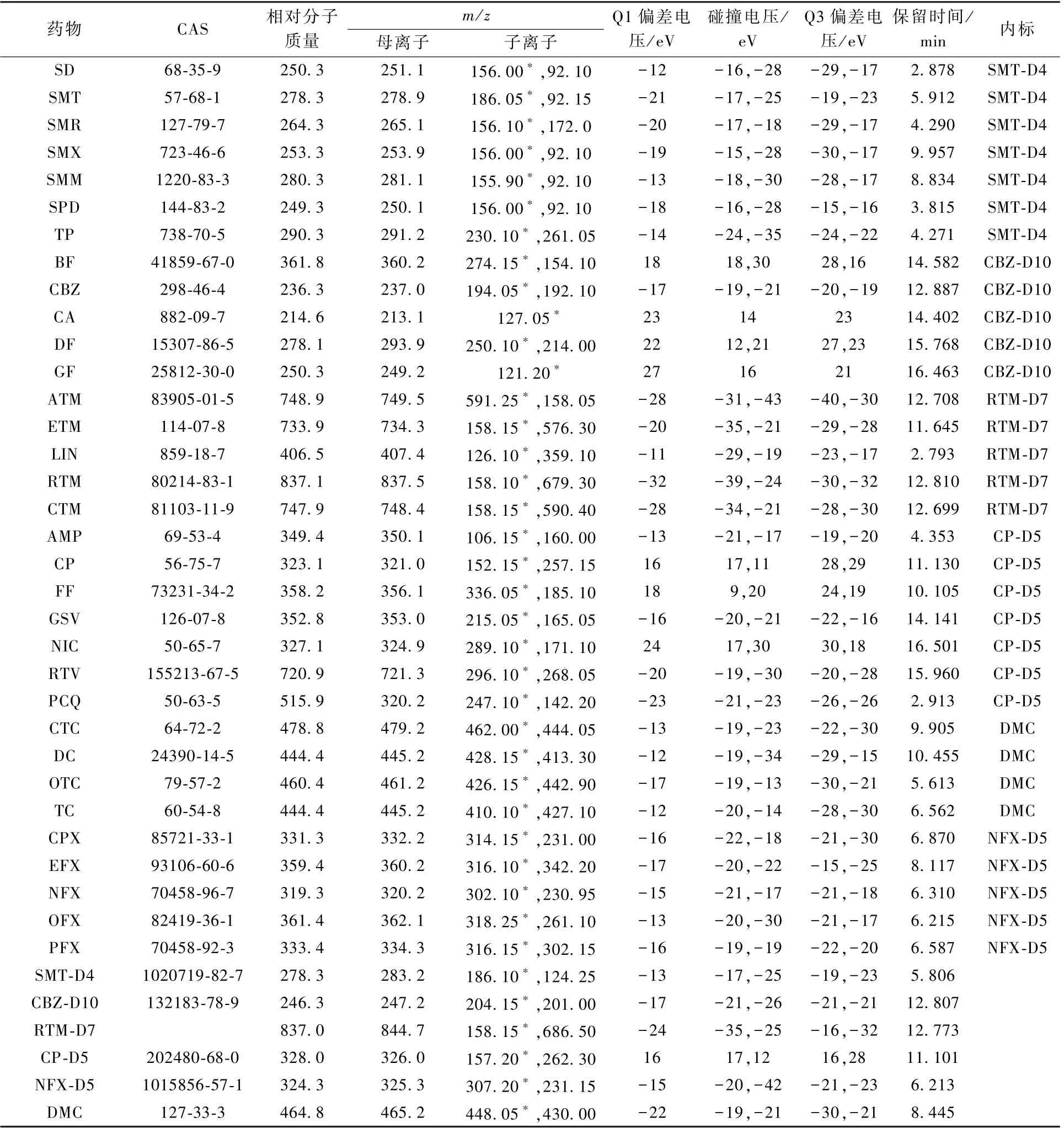
表1 33 種目標物的質譜參數、保留時間及內標Table 1 Parameters of mass spectrum,retention time and internal standard for the 33 target compounds
2.3 前處理條件的優化
在前期的研究和文獻報道的基礎上[12-13,25],本研究選取適用于酸性、堿性和中性化合物的通用型Oasis HLB 固相萃取柱對樣品進行富集和凈化。 不同藥物在HLB 固相萃取填料上的吸附程度與藥物本身的pKa和水樣pH 有關。 當酸性藥物的樣品pH 高于pKa或堿性藥物的樣品pH 低于pKa時,大多數藥物分子會以親水的離子形式存在,從而不容易被固相萃取柱保留[26]。 因此,本研究首先考察了樣品pH 對藥物回收率的影響。 用甲酸和氨水將水樣pH 分別調整為3.0、7.0、9.0,并向樣品中加入100 ng定量內標,進行加標回收實驗(10 m L 甲醇洗脫),每組3 個平行,加標量為100 ng/L,結果見圖2。 實驗結果表明,樣品pH 對藥物回收率結果影響較大。 CTC、DC、TC、OTC、AMP、SD在酸性條件下回收率較好,SMT、CPX、EFX、NFX、OFX、PFX、CA 在堿性條件下回收率較好,其他藥物的回收率在不同pH 條件下沒有明顯的差異。 為了簡化樣品分析的工作量,后續實驗中將CTC、DC、TC、OTC、AMP、SD 在pH=3 的條件下分析,其他藥物在pH= 9 的條件下分析。
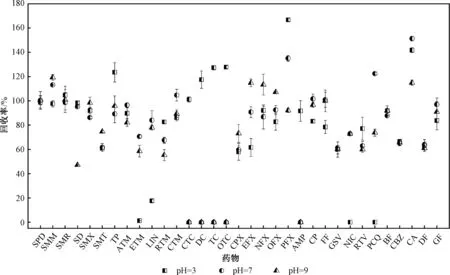
圖2 不同pH 條件下藥物的回收率Fig.2 Recoveries of pharmaceuticals under different pH condition of water samples
本研究中分析的目標物種類較多,理化性質各異,單一的洗脫溶劑無法獲得好的洗脫效果,首先對比了甲醇和乙腈對目標物的洗脫效果(洗脫溶劑用量10 m L),每組3 個平行,加標量為100 ng/L。 結果顯示,乙腈會嚴重降低喹諾酮類、大環內酯類藥物的回收率,回收率范圍為0 ~66.4%,而甲醇的洗脫效率明顯高于乙腈,因此初選甲醇作為洗脫溶劑。 根據文獻報道發現,向甲醇中加入甲酸或氨水,可以改變目標物在固相萃取柱上的存在形態,降低HLB 填料與目標物的相互作用力,增大回收率[27-28]。 因此,本研究對比了5%甲酸-甲醇、純甲醇和5%氨水-甲醇對目標物的洗脫效率(圖3)。 結果表明,對于酸性水樣,用5%甲酸-甲醇或5%氨水-甲醇作為洗脫溶劑的洗脫效率下降,特別對于四環素類抗生素影響較大,回收率范圍為90.0%~243%,而純甲醇對四環素類抗生素的洗脫效率明顯提升,這主要是由于甲醇的極性高,洗脫能力強,在甲酸、氨水存在的條件下洗脫能力反而變差,回收率降低[29]。 因此,在酸性條件下選擇純甲醇作為洗脫溶劑。 對于堿性水樣,5%氨水-甲醇作為洗脫溶劑會提升TP、ETM、RTM、CTM、CPX、PFX、GSV、NIC、CBZ、GF 的洗脫效率, 相比于甲醇溶劑分別提高了 3.55%、25.1%、35.4%、31.7%、43.8%、7.45%、13.9%、23.3%、7.97%、6.99%,但是會降低SMX、SMT、LIN、OFX、PCQ 的洗脫效率。 5%甲酸-甲醇作為洗脫溶劑與純甲醇的洗脫效率相比沒有明顯的提升。 因此,為了進一步提升藥物的洗脫效率,在堿性條件下首先用純甲醇進行洗脫,再用5%氨化甲醇進行洗脫。
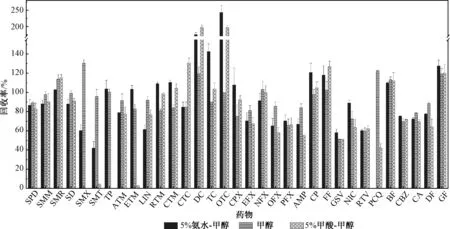
圖3 不同洗脫溶劑類型對藥物的回收率影響Fig.3 Effect of composition of elute solvent on the recoveries of pharmaceuticals
本研究還考察了洗脫溶劑用量對回收率的影響,pH=3 條件下分別選用8、10、12 m L 甲醇進行實驗。 pH= 9 條件下分別選用甲醇和體積分數5%氨化甲醇作為洗脫溶劑依次進行洗脫,兩者的體積比為1 ∶1,洗脫溶劑總體積分別選用8、10、12 m L,洗脫溶劑加入順序為先加入純甲醇,再加入體積分數5%氨化甲醇進行洗脫,每組3 個平行,加標量為100 ng/L,結果如圖4 所示。 實驗結果表明,無論是酸性水樣還是堿性水樣,提高洗脫溶劑的體積對于目標物的洗脫效率沒有明顯提升。 為了節省氮吹時間和溶劑用量,最終確定為8 m L 洗脫溶劑進行實驗。

圖4 洗脫溶劑用量對藥物回收率影響Fig.4 Effect of volume of elute solvent on the recoveries of pharmaceuticals
2.4 質量保證與質量控制
將混合標準儲備液(10 μg/m L)稀釋成0.05、0.10、0.20、0.50、1、5、10、50、100、200、250、500 ng/m L 的系列混合標準溶液,并分別向系列混合標準溶液中加入100 ng 內標進行測定。 以目標化合物的峰面積與對應的內標峰面積之比對質量濃度繪圖,得到每一個目標藥物的標準曲線,均呈現良好的線性關系(相關系數r>0.995)。 取地表水樣品為基底,加標濃度考慮低(10 ng)、中(100 ng)、高(500 ng)水平,利用優化的前處理結果對樣品進行處理分析,根據加標濃度計算加標回收率及相對標準偏差,加標回收率的計算如式(1)所示,結果見表2。 結果表明,在低、中、高3 個添加水平下(n = 6) 考察方法的回收率分別為55.0%~119%、53.7%~122%、62.7% ~116%,相對標準偏差分別為1.67% ~32.1%、1.75% ~18.8%、1.22%~12.3%。 以3 倍信噪比(S/N=3)對應的濃度為儀器檢出限(LOD), LOD 為0.001 ~1.71 ng/L。 以10 倍信噪比(S/N=10)對應的濃度為儀器定量限(LOQ),LOQ 為0.012 ~4.68 ng/L。 方法檢出限(MDL)根據各目標化合物的儀器檢出限、回收率和濃縮倍數等確定,其計算公式如式(2)所示[30]。 MDL 為0.011 ~7.60 ng/L,各目標物的質量控制參數見表2。

表2 方法檢出限、加標回收率、回歸方程及相關系數Table 2 Detection lim its,recoveries,calibration curve and correlation coefficient of the method
式中:R 為目標化合物的加標回收率,%;Ce為含有目標物的樣品經過前處理后的實測質量濃度,ng/L;C0為目標物在溶液中的質量濃度,ng/L。
式中M 為樣品濃縮倍數。
3 實際樣品分析
使用建立的方法對北京市涼水河12 個地表水樣中的藥物濃度進行分析,每種藥物的濃度水平如圖5 所示。 TC、OPX、RTV、DF 在涼水河的檢出率為100%,AMP 的檢出率為0%,其余藥物均有不同程度檢出,檢出率為8.33% ~91.7%。 磺胺類抗生素的平均質量濃度為14.9 ng/L,四環素類抗生素的平均質量濃度為5.70 ng/L,喹諾酮類抗生素的平均質量濃度為29.8 ng/L,大環內酯類抗生素的平均質量濃度為18.3 ng/L,非抗生素類藥物的平均質量濃度為5.90 ng/L。 OFX、TP、NFX 和CTM 的濃度水平較高,最高質量濃度分別為239、170、121 和84.3 ng/L。 AMP、SMM、GSV、SMT、OTC、CTC、PCQ、CP、PFX、NIC 和FF 的濃度水平較低,檢出范圍分別為ND(未檢出)、ND ~0.72 ng/L、ND ~1.54 ng/L、ND ~1.96 ng/L、ND ~2.19 ng/L、ND ~2.82 ng/L、ND ~2.84 ng/L、ND ~2.92 ng/L、ND ~3.15 ng/L、ND ~3.27 ng/L、ND ~4.48 ng/L。 值得注意的是,RTV 和PCQ 作為新冠肺炎診療方案中推薦的藥物,其中RTV 在涼水河中100%檢出,質量濃度為0.67 ~11.5 ng/L,這可能與疫情期間藥物的使用及其自身的持久性有關[18]。

圖5 北京市涼水河中33 種藥物的濃度水平Fig.5 The concentrations of 33 pharmaceuticals in the Liangshui River of Beijing
通過文獻分析發現(表3),涼水河地表水中磺胺類抗生素的濃度水平低于汾河和雄安新區境內河流濃度水平,與常州境內河流濃度水平相似,但要高于長江重慶段和潮汕境內河流濃度水平,這表明涼水河中磺胺類抗生素處于中等污染水平。 涼水河中大環內酯類抗生素ATM、ETM、LIN、RTM 的存在水平與其他地區相似,而CTM的質量濃度為ND ~84.3 ng/L,平均質量濃度為31.5 ng/L,略高于其他地區。 CTC、DC 在雄安新區和潮汕地區河流中未檢出,在汾河和涼水河中的濃度水平類似。 喹諾酮類藥物NFX、OFX 在河流中存在的濃度水平整體偏高,這可能與農田區有機肥料的施用有關,藥物通過地表徑流進入地表水,此外喹諾酮類藥物在環境中半衰期較長,導致其更易于在環境中累積[31-33]。AMP 在環境中的存在水平較低,涼水河中未檢出,雄安新區境內河流中檢出率為67.0%,質量濃度為ND ~2.32 ng/L。 涼水河中CP 和CBZ與文獻中報道的濃度水平類似。

表3 中國地表水中部分藥物濃度水平Table 3 The concentrations of some pharmaceuticals in su rface waters in China
據統計,武漢地區新冠疫情期間,洛匹那韋、利托那韋、磷酸氯喹、利巴韋林、阿比多爾、莫西沙星和甲潑尼龍7 種抗擊新冠病毒的藥物使用量約為2 472 kg(按確診50 333 人計算),其中檢測到利巴韋林在地表水中的質量濃度為1.04 ~52.2 ng/L[18],明顯高于本研究中報道的地表水中抗病毒類藥物濃度。 基于定量構效關系(QSAR)模型計算發現,抗病毒類藥物對水藻、水蚤、魚類和甲殼類動物具有生態毒性效應,其毒性效應與心血管藥物、抗焦慮藥、安眠藥、抗精神病藥、胃腸藥等相似[34-35]。 雖然目前對于地表水中抗病毒類藥物報道的濃度數據較少,但是隨著疫情的常態化發展,導致抗病毒藥物的使用量增加,地表水中抗病毒類藥物的存在水平、環境歸趨和生態風險應該引起足夠的重視。
4 結論
本研究采用SPE-HPLC-MS/MS 技術,通過重點優化目標分析物的色譜條件、質譜條件、樣品的pH、洗脫劑類型及用量等,建立了地表水中33 種藥物同時分析測定方法,方法檢出限為0.011 ~7.60 ng/L,地表水加標回收率為53.7%~122%,相對標準偏差為1.22%~32.1%(n=6)。 方法成功應用于北京市涼水河地表水藥物的分析,共檢出32 種藥物,檢出質量濃度為ND ~239 ng/L。抗生素類藥物在水體中的檢出質量濃度為ND ~239 ng/L,非抗生素類藥物的檢出質量濃度為ND ~54.6 ng/L。 其中,磺胺類抗生素的檢出質量濃度為ND ~47.2 ng/L,大環內酯類抗生素的檢出質量濃度為ND ~84.3 ng/L,四環素類抗生素的檢出質量濃度為ND ~25.3 ng/L,喹諾酮類抗生素的檢出質量濃度為ND ~239 ng/L,其他類抗生素的檢出質量濃度為ND ~4.47 ng/L,β-內酰胺類抗生素未檢出。 該研究結果為地表水典型藥物的分析提供了一種高效、準確的分析方法,具有較高的應用價值。
