鐵死亡在骨代謝功能細胞中的機制與展望
2024-01-12 13:13:44秦金然王亮王禮寧朱三木馬勇謝雁鳴魏戌章軼立
中國骨質疏松雜志 2023年12期
秦金然 王亮 王禮寧,2 朱三木 馬勇,2 謝雁鳴 魏戌 章軼立,2*
1.南京中醫藥大學中西醫結合學院,江蘇 南京 210023 2.江蘇省中醫退行性骨關節病臨床醫學創新中心,江蘇 無錫 214071 3.中國中醫科學院中醫臨床基礎醫學研究所,北京 100700 4.中國中醫科學院望京醫院,北京 100102
骨骼是人體中代謝相對活躍的組織,其正常生理功能與骨細胞、骨髓間充質干細胞、成骨細胞、破骨細胞等功能細胞的正常運轉有關。近年來,以骨質疏松癥為代表的骨代謝疾病發病率呈現逐年上升趨勢[1]。2018年國家衛生健康委發布的我國骨質疏松癥流行病學調查結果顯示,我國50歲以上人群的骨質疏松癥患病率為19.2%,65歲以上人群的骨質疏松癥患病率高達32.0%。隨著我國老齡人口數量的日益增加,骨質疏松癥患病率亦呈急速攀升趨勢,已成為政府、社會、個人亟待重視的公共健康問題[2-3]。
“鐵死亡”是由Brent R.Stockwell教授等于2012年首次提出,以細胞內活性氧(reactive oxidative species,ROS)堆積以及線粒體形態、膜電位改變為主要特征[4-5]。目前,關于鐵死亡的研究主要集中在腫瘤、神經退行性疾病、視網膜疾病、自身免疫性疾病等領域[6]。近年來,鐵死亡與骨質疏松癥為代表的骨代謝疾病研究取得了諸多進展。因此,本綜述擬從骨細胞、骨髓間充質干細胞、成骨細胞、破骨細胞等骨代謝功能細胞的角度,重點闡述鐵死亡發生機制及其與相關細胞的作用關系。
1 鐵死亡概述
與傳統的程序性細胞死亡(如細胞凋亡、壞死)方式不同,鐵死亡主要歸因于細胞內脂質過氧化、谷胱甘肽過氧化物酶4(glutathione peroxidase 4,GPX4)抗氧化系統失調以及鐵超負荷[7-8]。在形態學上,鐵死亡涉及細胞膜破裂和出泡,線粒體變小、膜密度增加、線粒體嵴減少甚至消失、線粒體外膜破裂,細胞核大小正常但缺少染色質凝聚[9]。
1.1 脂質過氧化
GPX4是鐵死亡的關鍵調節因子,其通過將高反應性脂質氫過氧化物還原為非反應性脂質醇來保護細胞免受膜脂質過氧化并維持氧化還原穩態[10]。
根據烴鏈飽和度的差異,脂肪酸可分為三類:飽和脂肪酸(saturated fatty acids,SFAs)、單不飽和脂肪酸(monounsaturated fatty acids,MUFAs)和多不飽和脂肪酸(polyunsaturated fatty acids,PUFAs)[11]。含有PUFAs的磷脂極易發生過氧化,促進 PUFAs 摻入膜磷脂可以促進鐵死亡;反之,增加膜磷脂中氧化性較低的MUFAs數量可以有效抵抗鐵死亡[12]。因此,控制PUFAs和MUFAs代謝的酶和通路在確定細胞對鐵死亡敏感性方面起關鍵作用。Mortensen等[13]認為PUFAs在鐵死亡過程中占據重要地位,因為它們傾向于形成過氧自由基,通過鏈式反應在整個膜中傳播,繼而導致不可挽回的膜損傷和細胞死亡。而脂氧合酶(lipoxygenases,LOXs)和磷酸化酶激酶G2(phosphorylase kinase gamma 2,PHKG2)是鐵死亡過程中脂質過氧化的兩個關鍵驅動因素。脂氧合酶的過表達使細胞對鐵死亡敏感[14]。PHKG2可調節脂氧合酶對鐵的可利用性,并且脂氧合酶通過雙烯丙基位置的多不飽和脂肪酸過氧化促使鐵死亡[15]。此外,相關學者還發現脂氧合酶通過PHKG2依賴性鐵池氧化PUFAs是鐵死亡所必需的,并且GPX4中催化硒化半胱氨酸的共價抑制阻止了PUFAs氫過氧化物的消除,這些發現為在不同情況下調控鐵死亡提供了新的研究思路[16]。
1.2 GPX4抗氧化系統失調
氧化應激引起不受控制的脂質過氧化物產生通常會導致線粒體脂質過氧化和損傷,繼而引發鐵死亡。脂質過氧化物是脂質過氧化過程中的關鍵中間體,脂質氫過氧化物酶GPX4將脂質氫過氧化物蛻變為脂質醇,此過程可防止鐵(Fe2+)依賴性形成有毒脂質ROS。GPX4活性的丟失及隨后脂質氫過氧化物的積累會導致鐵死亡,既往研究表示GPX4是鐵死亡發生過程中的中樞調節因子,其可以在復雜的細胞膜環境中加速脂質過氧化物的衰減[17]。
鐵死亡誘導因子可通過多種途徑直接或間接的影響谷胱甘肽過氧化物酶(glutathione peroxidase,GPX),導致抗氧化能力下降和細胞中脂質活性氧積累,最終導致氧化細胞死亡。erastin和RSL3是最早的GPX系統抑制劑,具有不同的細胞死亡觸發機制。erastin通過抑制系統XC-阻止胱氨酸的攝取,從而消耗谷胱甘肽(glutathione,GSH),發揮間接抑制GPX4的作用;而RSL3則是直接滅活GPX4,誘導鐵死亡[18]。與之前描述的鐵死亡誘導劑不同,Gaschler等[19]研究發現FINO2可啟動多管齊下的鐵死亡誘導機制。Shimada等[20]發現CIL56和FIN56催化的細胞死亡常常伴隨著脂質ROS的產生,且維生素E和鐵螯合劑對這些化合物誘導的細胞死亡具有抑制作用,表明它們可能是鐵死亡誘導劑。此外,作者還證實了 FIN56通過誘導GPX4降解來誘導鐵死亡。
1.3 鐵代謝紊亂
鐵離子對于細胞生長、氧氣利用以及各種酶的活性等諸多的細胞和分子過程至關重要,反之,過量的鐵離子會通過推動有毒自由基的產生繼而致使細胞功能障礙,異常的鐵積累可能會導致多種器官損傷[21]。細胞鐵超負荷可降低細胞活力、超氧化物歧化酶和GSH水平,增加了ROS產生、脂質過氧化、丙二醛水平以及鐵死亡相關蛋白的表達,并誘導線粒體的超微結構變化[22]。大量Fe3+在金屬還原酶的作用下還原成Fe2+,游離Fe2+氧化性強,易于與H2O2發生芬頓反應,產生羥基自由基,繼而促進脂質過氧化反應發生損傷細胞膜致使細胞死亡[23]。
Geng等[24]通過體外實驗證明敲低膜鐵轉運蛋白(ferroportin,FPN)會增加鐵依賴性脂質ROS積累繼而加速erastin誘導的鐵死亡。后續的相關研究再次驗證了這一結果,并提示FPN過表達會抑制erastin誘導的鐵死亡[25]。
2 鐵死亡與骨代謝功能細胞
2.1 鐵死亡與骨細胞
一直以來,骨細胞被認為是成骨細胞和破骨細胞傳遞調節信號的“指揮官”,在控制骨重塑中發揮關鍵作用。Ma等[26]認為骨細胞是預防和治療鐵超負荷引起的骨質疏松癥的主要靶點,在指定濃度的檸檬酸鐵胺(ferric ammonium citrate,FAC)處理24 h后,FAC以劑量依賴的方式顯著增加骨硬化蛋白和RANKL的表達水平,同時降低OPG的表達水平,導致RANKL/OPG的表達水平升高,而使用去鐵胺(Deferoxamine,DFO)和N-乙酰半胱氨酸(N-acetylcysteine,NAC)在處理FAC干預的骨細胞可逆轉該結果。研究發現鐵超負荷不僅會破壞骨細胞微絲骨架,并且會導致骨細胞中微絲骨架的分布改變;此外,鐵超負荷顯著阻礙MLO-Y4細胞系的細胞活力,同時通過上調骨細胞中RANKL的表達和分泌刺激破骨細胞分化[27]。糖尿病微環境可顯著促進骨細胞鐵死亡,主要表現為大量的脂質過氧化、鐵超負荷以及鐵死亡途徑的異常激活。靶向鐵死亡(注射鐵死亡抑制劑Fer-1)或 HO-1(注射HO-1抑制劑ZnPP)通過破壞脂質過氧化和HO-1激活之間的惡性循環,有效減少糖尿病骨質疏松癥(diabetic osteoporosis,DOP)中的骨細胞死亡,最終減少骨質流失[28]。Sun等[29]的研究結果表明地塞米松(DEX)可上調p53以抑制溶質載體家族7成員11(solute carrier family 7 member 11,SLC7A11)/GPX4的表達,從而誘導MLO-Y4細胞(小鼠骨樣細胞)鐵死亡。
2.2 鐵死亡與骨髓間充質干細胞
骨髓間充質干細胞(bone mesenchymal stem cells,BMSCs)是一種多能干細胞,其在正常骨代謝中起關鍵作用,被認為是具有維持和修復骨組織潛力的種子細胞[30-31]。近年來,隨著對鐵死亡研究的深入,人們發現可通過PI3K/AKT/mTOR信號通路抑制鐵死亡來改善BMSCs的抗氧化應激過程[32]。Li等[33]的實驗結果顯示地塞米松誘導BMSCs鐵死亡,且褪黑素可通過PI3K/AKT/mTOR途徑減少地塞米松誘導的BMSCs鐵死亡。
Engeletin(二氫山奈酚 3-鼠李糖苷)是一種從水果和蔬菜中分離出來的天然化合物[34]。研究發現Engeletin對BMSCs的成骨能力具有積極影響,此外還可以通過Nrf2/Keap1途徑減輕erastin誘導的氧化應激損傷,抑制BMSCs鐵死亡[35]。Liu等[36]使用erastin誘導BMSC鐵死亡,發現并首次證明了NOP2/Sun RNA甲基轉移酶5(NOP2/Sun RNA methyltransferase 5,NSUN5)通過與腫瘤壞死因子受體相關蛋白(tumor necrosis factor receptor associated protein 1,TRAP1)相互作用以及鐵蛋白重鏈1(ferritin heavy chain 1,FTH1)和鐵蛋白輕鏈(ferritin light chain,FTL)mRNA的5-甲基胞嘧啶(5mC)修飾來抑制BMSCs的鐵死亡。由此可見,NSUN5-FTH1/FTL通路組分的治療靶點具有提高BMSCs存活的潛力。
Song等[37]發現,FA互補基團D2(FA complementation group D2,FANCD2)可以通過減少鐵超負荷和脂質過氧化,抑制erastin刺激的BMSCs鐵死亡。并且ROS的過度積累也會激活G蛋白軸并破壞氧化平衡穩態以誘導BMSCs中的鐵死亡。Perillo等[38]認為p53通過在中度氧化應激下阻止ROS的過度增加來促進細胞存活,而當氧氣增加超過閾值水平時,它則會轉變為ROS誘導劑,觸發細胞死亡。高糖環境中,BMSCs的成骨細胞分化與細胞增殖活性被顯著抑制,致使細胞內ROS和LPO水平增高,而鐵死亡抑制劑Fer-1可恢復其正常[39]。除此之外,已有研究表明由FAC引起的細胞內鐵超負荷可以通過下調Wnt靶基因Lef1、Bmp4、Smad6 和細胞周期蛋白D1(Cyclin D1)的表達來提高鐵死亡敏感性,從而抑制間充質干細胞向成骨細胞分化[40]。最后,維生素K2(VK2)也已被證實可通過腺苷酸活化蛋白激酶(AMP-activated protein kinase,AMPK)/SIRT1信號通路抑制BMSC鐵死亡,從而緩解2型糖尿病骨質疏松癥(type 2 diabetes osteoporosis,T2DOP)[41]。
2.3 鐵死亡與成骨細胞
成骨細胞是BMSCs的終末分化產物,調節骨的形成以及重建過程。鐵是眾多生物過程的必需微量元素,在成骨細胞的分化和礦化過程中起關鍵作用。在相似的條件下,為實現適當的分化和礦化,成骨細胞比脂肪細胞有更高的鐵需求[42],但鐵超負荷同樣會抑制成骨作用,導致成骨細胞鐵死亡[43]。
鐵轉運蛋白(ferroportin1,FPN1)是目前發現的哺乳動物中唯一的細胞中鐵外排蛋白,是鐵死亡信號通路的關鍵蛋白[44-45]。FPN1通過提高細胞內ROS水平,引起氧化應激,從而導致細胞鐵死亡。付殷等[46]的研究表明,淫羊藿苷可以通過抑制FPN1過表達調節鐵死亡信號通路,最終提高成骨細胞增殖、礦化能力。根據前期研究可知,AKT/PI3K信號通路是miR-483-5p-SATB2軸在OVX大鼠中作用的下游靶標,通過促進骨髓干細胞的增殖和遷移改善骨質疏松癥的發展[47]。Hao等[48]驗證了ATM和AKT/PI3K通路可改善成骨功能并抑制成骨細胞鐵死亡。此外,Luo等[40]在機制上揭示了鐵劑量依賴性抑制Wnt信號傳導。Wnt 激動劑、鐵死亡抑制劑或抗氧化劑褪黑激素可逆轉鐵被抑制的Wnt信號傳導,通過減少ROS和脂質過氧化物(lipid peroxidation,LPO)的產生恢復成骨細胞分化,從而在不降低鐵超負荷的情況下顯著預防鐵死亡。另有研究證實,DEX通過p53/SLC7A11/GPX4通路誘導MC3T3-E1細胞鐵死亡[29]。
高糖可通過積累RAS以及消耗細胞內的GSH來誘導MC3T3-E1成骨細胞的鐵死亡[49]。Zhao等[50]的研究表明高糖可誘導轉錄激活因子3(activation transcription factor 3,ATF3)上調,通過抑制ATF3的功能可增加GPX4水平并減少ROS和脂質過氧化物的積累,從而達到抑制成骨細胞鐵死亡的目的。線粒體鐵蛋白(mitochondrial ferritin,FtMt)的過表達可降低高糖條件下成骨細胞發生鐵死亡,并且FtMt下調可通過ROS/PTEN誘導假定激酶1(PTEN-induced putative kinase 1,PINK1)/帕金森病蛋白(parkinsons disease protein,Parkin)通路誘導線粒體自噬,促進成骨細胞鐵死亡[49]。晚期糖基化終產物(advanced glycation end products,AGEs)是在高糖條件下形成的一組具有破壞潛力的修飾蛋白質和(或)脂質[51]。Ge等[52]在研究中首次揭示AGEs通過誘導鐵死亡顯著抑制了成骨細胞的增殖、分化和礦化。研究表明高脂肪環境同樣會抑制成骨細胞增殖和成骨分化,鐵死亡的相關指標隨之發生顯著變化,并且發現鐵死亡抑制劑可抑制由于高脂肪飲食導致的小鼠骨質流失的發展[53]。MaR1(Maresin1)是一種由多不飽和脂肪酸產生的內源性促消退脂質介質,其在體內外通過激活核因子E2相關因子2(nuclear factor erythroid 2-related factor 2,Nrf2)抑制高葡萄糖誘導的成骨細胞鐵死亡,緩解T2DOP[54]。激活METTL3/凋亡信號調節激酶1(apoptosis signal-regulating kinase 1,ASK1)-p38信號通路可能是高糖高脂肪誘導成骨細胞鐵死亡繼而導致糖尿病骨質疏松癥的主要原因之一[55]。ASK1-p38信號通路是一個與鐵離子濃度密切相關的信號轉導途徑,研究發現鐵超負荷可以通過調控ASK1-p38信號通路調控成骨細胞鐵死亡[56]。最后,研究發現褪黑素可通過激活Nrf2/HO-1信號通路,顯著降低MC3T3-E1的鐵死亡水平,提高成骨能力[57]。
2.4 鐵死亡與破骨細胞
破骨細胞是體內唯一具有骨吸收能力的細胞。據報道,骨髓來源巨噬細胞(bone marrow derived macrophage,BMM)中產生的ROS會增強RANKL誘導破骨細胞分化的能力[58]。有研究表明,過量的鐵進入細胞內會通過芬頓反應產生ROS,并進一步激活細胞內絲裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)信號通路。ROS/MAPKs/核轉錄因子-κB (nuclear factor-kB,NF-kB)/核苷酸結合寡聚化結構域樣受體蛋白3(nod-like receptor protein 3,NLRP3)的激活,可促進糖尿病性骨質疏松中破骨細胞介導的骨丟失[59]。Ni等[60]研究也證實了鐵死亡參與RANKL誘導的破骨細胞分化,并且在該過程中鐵饑餓反應和正常氧濃度下的鐵蛋白吞噬有助于RANKL誘導的破骨細胞鐵死亡;此外,作者還證明了體內HIF-1α可抑制通過鐵蛋白吞噬誘導的鐵死亡發生。Qu等[61]使用RANKL誘導的細胞模型評估唑來膦酸(ZA)對破骨細胞功能的調節作用,結果顯示鐵離子、ROS以及MDA含量增加,GPX4和GSH水平降低,由此可見,ZA處理抑制了破骨細胞的細胞活力,促進破骨細胞鐵死亡。
鐵調素是一種富含半胱氨酸的小分子肽,其由肝細胞產生,可抑制FPN活性[62],是鐵代謝和鐵穩態的關鍵調節劑,它受到鐵超負荷和炎癥的刺激,可以通過利用鐵調素來增加鐵的積累[63]。鐵可以增加破骨細胞的脂質過氧化,并誘導鐵死亡,從而抑制破骨細胞分化[64]。Li等[65]的研究認為過量的鐵通過增加TNF-α的分泌來破壞小鼠的骨承重能力,且TNF-α會促進破骨細胞的分化并增強骨吸收。
在破骨細胞分化過程中,破骨細胞通過轉鐵蛋白受體1(transferrin receptor 1,TfR1)介導的轉鐵蛋白(transferrin,Tf)內吞作用吸收大量鐵。Jin等[64]發現Tf能增強青蒿琥酯對破骨細胞的活力和分化的抑制作用,作者認為其機制與青蒿琥酯誘導破骨細胞鐵死亡有關。
鑒于鐵死亡在不同骨代謝功能細胞涉及的關鍵分子、信號通路、外部影響因素、組織局部微環境等不同,相關總結信息見表1、圖1。
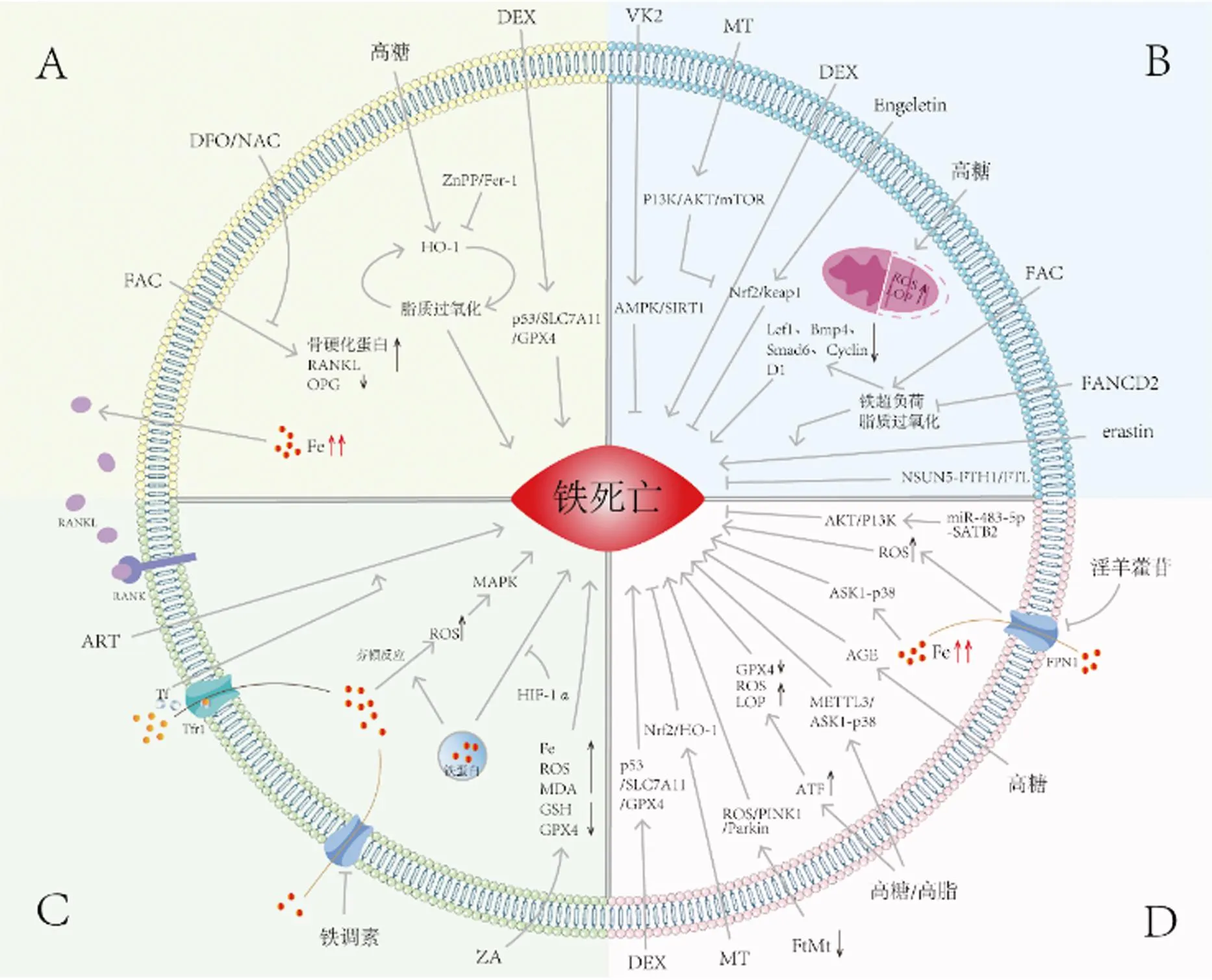
圖1 鐵死亡在骨代謝功能細胞中的作用機制Fig.1 Mechanism of action of ferroptosis in functional cells of bone metabolism注:A:骨細胞;B:骨髓間充質干細胞;C:破骨細胞;D:成骨細胞;ZnPP:HO-1抑制劑;Fer-1:鐵死亡抑制劑;FAC:檸檬酸鐵胺;DFO:去鐵胺;NAC:N-乙酰半胱氨酸;SLC7A11:溶質載體家族7成員11;DEX:地塞米松;FtMt:線粒體鐵蛋白;VK2:維生素K2;MT:褪黑素;Engeletin:二氫山奈酚 3-鼠李糖苷;FANCD2:FA互補基團D2;erastin:鐵死亡誘導劑;AGE:晚期糖基化終產物;ROS:活性氧;LOP:脂質過氧化物;GPX4:谷胱甘肽過氧化物酶4;FPN1:鐵轉運蛋白1;PINK1:PTEN誘導假定激酶1;Parkin:帕金森病蛋白;MAPK:絲裂原活化蛋白激酶;ZA:唑來膦酸;GSH:谷胱甘肽;ART:青蒿琥酯;Tf:轉鐵蛋白;Tfr1:轉鐵蛋白受體1;MDA:丙二醛;RANK:核因子-κB受體活化體;RANKL:核因子-κB受體活化體配體;OPG:骨保護素。

表1 骨質疏松癥相關細胞與鐵死亡之間的聯系Table 1 Association between osteoporosis-associated cells and ferroptosis
3 總結與展望
越來越多的證據表明,鐵死亡參與骨代謝疾病的病理生理過程,通過抑制或刺激這種獨特的細胞死亡方式來干預骨質疏松癥為代表的骨代謝疾病成為未來研究的重點。因此,持續關注鐵死亡在目標疾病發生發展中的作用,或可為開發新的治療策略提供希望。
