超高效液相色譜-串聯質譜法同時測定復雜基質保健食品中7種脂溶性維生素營養補充劑的含量
2024-03-18 14:54:28余家勝沈珊珊楊佳雯趙志紅
理化檢驗-化學分冊 2024年2期
吳 琴, 余家勝, 李 瑞, 沈珊珊, 楊佳雯, 趙志紅
(杭州娃哈哈集團有限公司 浙江省食品生物工程重點實驗室, 杭州 310018)
維生素是維持人和動物正常生理功能必不可缺的低相對分子質量有機化合物,根據其物理特性可以分為水溶性維生素和脂溶性維生素。其中,脂溶性維生素主要包括維生素A(VA)、維生素D(VD)、維生素E(VE)、維生素K(VK)[1]。維生素A存在于動物性食品中,主要來源為視黃醇酯[2-3]。維生素E極易被氧化,是良好的脂溶性抗氧化劑[4-6]。缺乏維生素D會引起骨軟化等疾病[7-8]。維生素K有K1(VK1)、K2(VK2)兩種,維生素K1主要存在于綠葉蔬菜和一些食用油中[9],維生素K2主要由發酵食品中的細菌產生[10]。
不同脂溶性維生素的化學性質存在一定差異,同時測定面臨許多問題和挑戰。提取維生素A、維生素D、維生素E的方法通常為皂化法[11],但是該方法不能用于皂化維生素K1和K2,因為這兩種物質易在堿性條件下降解。國際公認的提取嬰幼兒配方乳粉和成人/兒童營養食品中維生素A、維生素E的方法是先酶解,再用甲醇沉淀蛋白,最后用溶劑提取[12-13],且這種方法可用于提取維生素K1[14];提取食品和補充劑中的維生素D的方法仍為皂化法[15-16]。
隨著消費者對保健食品風味要求的提高,其基質也越來越復雜,因此建立一種準確、快捷、經濟的檢測復雜基質保健食品中脂溶性維生素的方法很有必要。本工作采用酶解法降解復雜基質保健食品中的脂肪、蛋白質,利用弱堿性乙醇溶液除去降解雜質,正己烷提取7種脂溶性維生素營養補充劑[維生素A乙酸酯、維生素A棕櫚酸酯、維生素D2(VD2)、維生素D3(VD3)、DL-α-維生素E乙酸酯、維生素K1和維生素K2],并參考文獻[17-18]采用超高效液相色譜-串聯質譜法同時測定這7種脂溶性維生素營養補充劑的含量,該方法前處理簡單,不需要復雜的皂化反應和固相萃取小柱凈化,即可實現7種脂溶性維生素營養補充劑的同時測定。
1 試驗部分
1.1 儀器與試劑
ACQUITY UPLC-Xevo TQ MS型超高效液相色譜-三重四極桿質譜儀,附Masslynx質譜軟件;R-210型旋轉蒸發儀;XS205DU型電子天平;AS-MTV100-114型多管渦旋振蕩器;S470-K型pH計;WB 22型恒溫水浴振蕩器;XMTD-608型氮氣吹掃儀。
單標準儲備溶液:100 mg·L-1,取適量各脂溶性維生素營養補充劑標準品或標準溶液,用甲醇溶解并定容至10 mL棕色容量瓶中,搖勻備用。
基質匹配單標準中間液:10 mg·L-1,取適量單標準儲備溶液,用空白樣品溶液稀釋,搖勻備用。
基質匹配混合標準溶液系列:取適量基質匹配單標準中間液,用空白樣品溶液稀釋,配制成維生素A乙酸酯、維生素A棕櫚酸酯、DL-α-維生素E乙酸酯的質量濃度為50,100,200,500,1 000 μg·L-1,維生素D2、維生素D3、維生素K1和維生素K2的質量濃度為10,20,40,100,200 μg·L-1的基質匹配混合標準溶液系列。
維生素K1(CAS號84-80-0)標準品的純度為99.6%;維生素K2(CAS號2124-57-4)標準溶液的質量濃度為100 mg·L-1;DL-α-維生素E乙酸酯(CAS號7695-91-2)標準品的純度為98.8%;維生素A乙酸酯(CAS號127-47-9,)標準品的純度為99.8%;維生素A棕櫚酸酯(CAS號79-81-2)標準品的純度為98.7%;維生素D3(CAS號67-97-0)標準品的純度為99.7%;維生素D2(CAS號50-14-6)標準品的純度為99.4%;脂肪酶的酶活力不小于30 U·mg-1;木瓜蛋白酶的酶活力不小于10 U·mg-1。
無水乙醇、碳酸鉀、正己烷、石油醚、乙醚均為分析純;甲醇、甲酸均為色譜純;試驗用水為實驗室一級用水。
1.2 儀器工作條件
1.2.1 色譜條件
Waters Acquity UPLC HSS T3 色譜柱(100 mm×2.1 mm,1.8 μm);柱溫35 ℃;流動相A為0.1%(體積分數,下同)甲酸溶液,B為含0.1%甲酸的甲醇溶液;流量0.3 μL·min-1;進樣量5 μL。梯度洗脫程序:0~3.0 min時,B由85%升至100%,保持5.5 min;8.5~9.0 min時,B由100%降至85%,保持1.0 min。
1.2.2 質譜條件
電噴霧離子(ESI)源,正離子(ESI+)模式;離子源溫度150 ℃;毛細管電壓4.00 kV;脫溶劑氣溫度200 ℃;脫溶劑氣流量800 L·h-1,錐孔氣流量50 L·h-1;多反應監測(MRM)模式。其他質譜參數見表1,其中“*”代表定量離子。
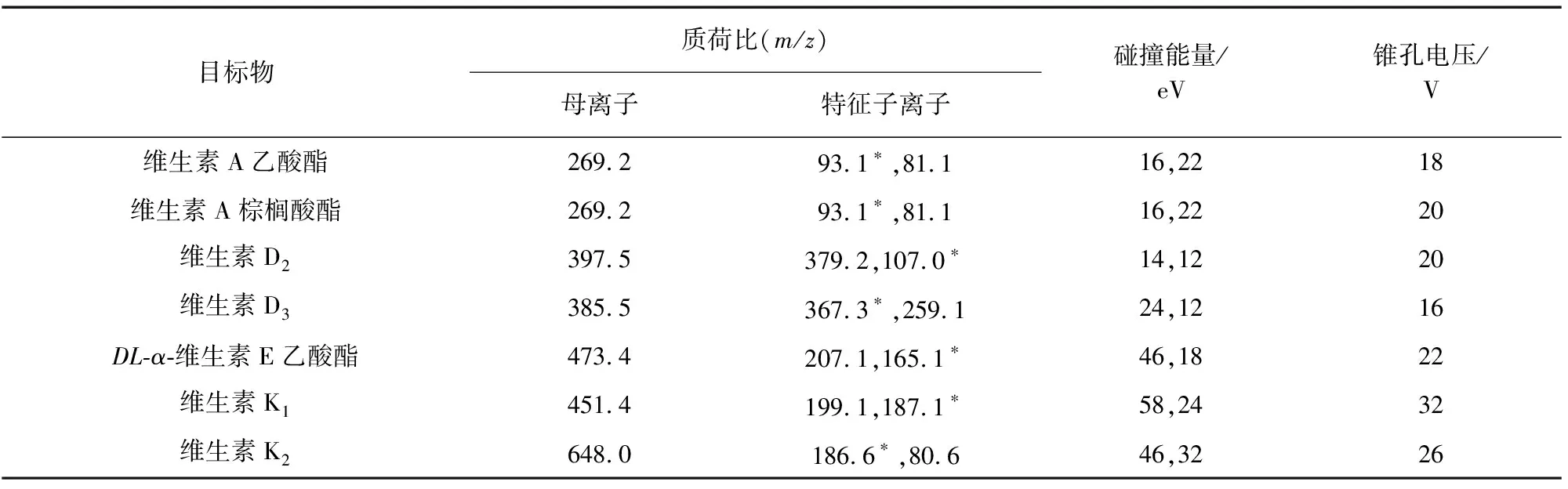
表1 質譜參數Tab. 1 MS parameters
1.3 試驗方法
將樣品混勻,分取約1 g置于50 mL離心管中,加入約50 ℃溫水5 mL和磷酸鹽緩沖液(pH 7.0) 5 mL,混勻后加入0.3 g脂肪酶、0.5 g木瓜蛋白酶,加蓋渦旋2~3 min,置于37 ℃恒溫水浴振蕩器中振蕩120 min。取出酶解好的樣品溶液,加入10 mL無水乙醇和1 g碳酸鉀,混勻后加入10 mL正己烷和10 mL水,渦旋或振蕩提取10 min,以6 000 r·min-1轉速離心5min,上層相轉移至100 mL旋蒸瓶中。在下層相中加入10 mL正己烷,重復上述提取和離心操作一次。合并上層相至旋蒸瓶中,于40 ℃旋蒸至近干,殘液用氮氣吹干,用甲醇溶解殘渣并定容至5 mL容量瓶中,搖勻后過0.22 μm濾膜,濾液上機檢測。對于目標物檢出量超出線性范圍的樣品,需要先稀釋再測定。
2 結果與討論
2.1 基質的影響及消除
復雜基質保健食品,如本工作研究的顆粒型保健食品,其中除了有維生素等營養補充劑外,還添加了大量的可可粉、全脂乳粉等進行調味,脂類和蛋白類化合物含量較高。若采用有機溶劑直接提取,共存脂肪、蛋白質也會被提取到提取液中,不僅影響維生素的測定,還會降低質譜響應值和縮短儀器使用壽命。本工作參考國家標準GB 5009.158-2016《食品安全國家標準 食品中維生素K1的測定》[19],先采用酶解法降解脂肪和蛋白質,酶解完成后加入無水乙醇、碳酸鉀、正己烷,使酶解產生的脂肪酸和甘油等降解雜質溶于弱堿性的乙醇溶液中,而目標物則留在了正己烷相,從而完成雜質的分離和目標物的提取。
2.2 酶解反應條件的選擇
由相關文獻可知,脂肪酶酶活性可在pH 6.5~9.0、溫度25~40 ℃的條件下維持在較高的水平[20];木瓜蛋白酶酶活性在pH 6.8~7.6內較高,在70 ℃以上才降低[21]。因此,試驗選擇在pH 7.0,溫度37 ℃條件下進行酶解反應。
參考本課題組前期研究[22],以空白加標樣品(維生素A乙酸酯、維生素A棕櫚酸酯和DL-α-維生素E乙酸酯的加標量為2.5 μg·g-1,維生素D2、維生素D3、維生素K1和維生素K2的加標量為0.5 μg·g-1)為待測對象,固定脂肪酶用量為0.3 g,考察了木瓜蛋白酶用量分別為0.1,0.3,0.5,0.8,1.0 g時各目標物的回收率,結果見圖1。
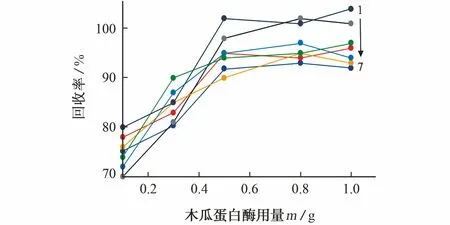
1-DL-α-VE乙酸酯;2-VK1;3-VA棕櫚酸酯;4-VD3;5-VA乙酸酯;6-VK2;7-VD2
由圖1可知,木瓜蛋白酶的用量不小于0.5 g時,各目標物的回收率較高且基本保持不變,因此試驗選擇的木瓜蛋白酶的用量為0.5 g。
2.3 提取溶劑的選擇
脂溶性維生素可被多種有機溶劑提取,相關國家標準采用體積比1∶1的石油醚-乙醚混合溶液提取皂化溶液中的維生素A、維生素D、維生素E[23],采用正己烷提取維生素K1[19];國家衛生和計劃生育委員會相關公告采用異丙醇提取維生素K2[24]。本工作涉及的脂溶性維生素種類較多,因此比較了上述3種提取溶劑對7種目標物回收率的影響,結果見圖2。
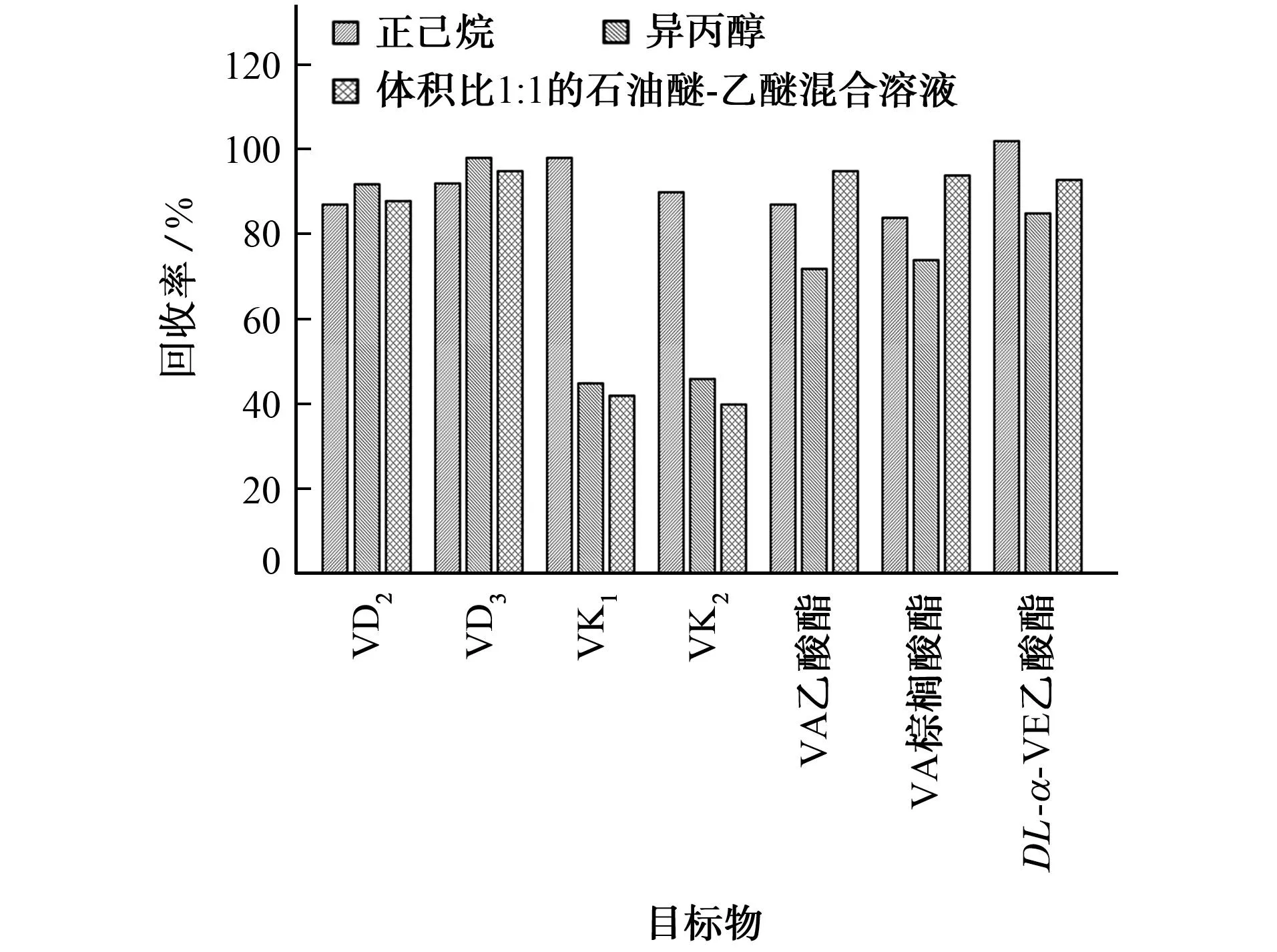
圖2 提取溶劑對目標物回收率的影響Fig. 2 Effect of extraction solvent on the recovery of targets
由圖2可知:以體積比1∶1的石油醚-乙醚混合溶液提取時,維生素A乙酸酯和維生素A棕櫚酸酯的回收率較大,但是維生素K1和維生素K2的回收率較小;以異丙醇提取時,維生素D2和維生素D3的回收率較大,但是維生素K1和維生素K2的回收率仍舊較小;以正己烷提取時,各目標物均獲得較高的回收率,其中維生素K1和維生素K2的回收率顯著增大。綜合考慮回收率以及石油醚、乙醚、異丙醇在使用過程中對試驗人員健康影響較大等因素,試驗選擇的提取溶劑為正己烷。
2.4 電離模式的選擇
ESI+和大氣壓化學電離正離子(APCI+)模式都可用來電離脂溶性維生素[25]。為確定最優電離模式,在20 μL·min-1流量下用蠕動泵進樣10 ng各目標物,分別用ESI+和APCI+模式電離。結果顯示,兩種電離模式下維生素A乙酸酯、維生素A棕櫚酸酯、維生素D2、維生素D3、DL-α-維生素E乙酸酯、維生素K1和維生素K2母離子質譜響應值比值分別為4.7,6.3,1.1,1.0,5.4,21.4,22.2,說明:對于維生素K1和維生素K2,ESI+模式比APCI+模式更靈敏;對于維生素D2和維生素D3,兩種模式電離效果基本一致;對于維生素A和維生素E,ESI+模式略優。在實際檢測時,相關樣品中維生素A和維生素E含量較高,且APCI+模式電離后基線噪聲更低,兩種電離模式都可選用;但是維生素K1和K2含量相對較低,應選擇靈敏度更高的ESI+模式。綜合考慮,試驗選擇的電離模式為ESI+。
2.5 色譜柱的選擇
試驗比較了HSS T3色譜柱(100 mm×2.1 mm,1.8 μm)、BEH C18色譜柱(100 mm×2.1 mm,1.7 μm)、HSS C18色譜柱(100 mm×1.7 mm,1.8 μm)等3種色譜柱對7種目標物的分離效果。結果顯示,HSS T3色譜柱(100 mm×2.1 mm,1.8 μm)所得各目標物響應值更高,分離度更好,峰形更尖銳,因此試驗選擇的色譜柱為HSS T3色譜柱(100 mm×2.1 mm,1.8 μm)。
在優化的儀器工作條件下,各目標物的定量離子色譜圖見圖3。
圖3 目標物定量離子的色譜圖Fig. 3 Chromatograms of quantitative ions of targets
2.6 工作曲線、檢出限和測定下限
按照儀器工作條件測定基質匹配混合標準溶液系列,以各目標物的質量濃度為橫坐標,其對應的峰面積為縱坐標繪制工作曲線。結果顯示,維生素A乙酸酯、維生素A棕櫚酸酯、DL-α-維生素E乙酸酯的質量濃度均在50~1 000 μg·L-1內,維生素D2、維生素D3、維生素K1和維生素K2的質量濃度均在10~200 μg·L-1內與各自對應的峰面積呈線性關系,其他線性參數見表2。
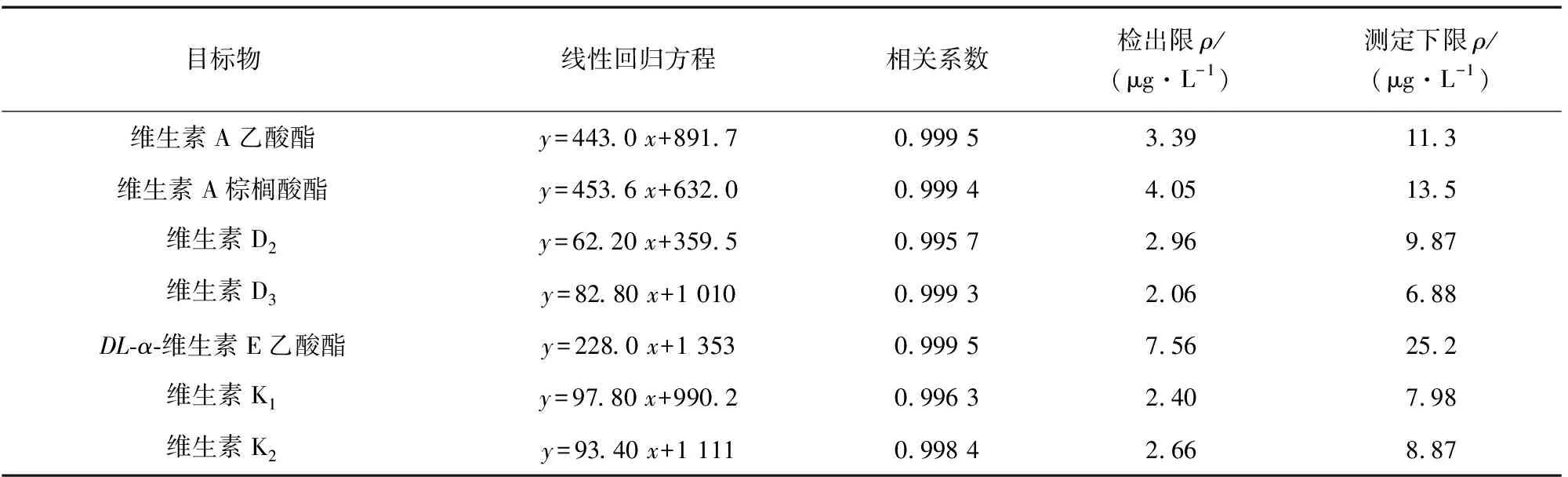
表2 線性參數、檢出限和測定下限Tab. 2 Linearity parameters, detection limits and lower limits of determination
以3倍信噪比(S/N)計算檢出限(3S/N),以10倍信噪比計算測定下限(10S/N),結果見表2。
2.7 精密度與回收試驗
按照試驗方法對空白樣品進行低、中、高等3個濃度水平的加標回收試驗,每個濃度水平平行測定6次,計算回收率和測定值的相對標準偏差(RSD),結果見表3。
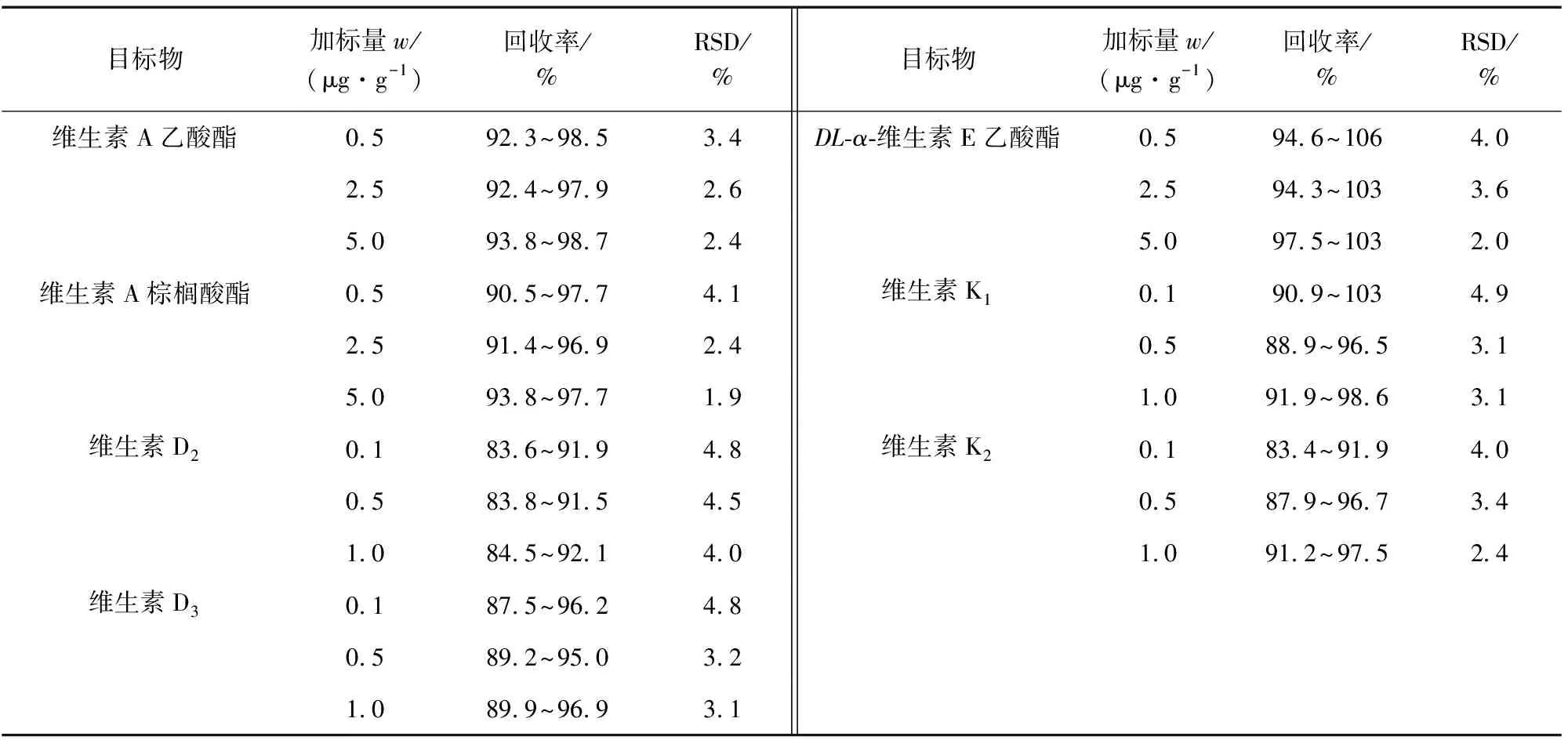
表3 精密度和回收試驗結果(n=6)Tab. 3 Results of tests for precision and recovery(n=6)
由表3可知,各目標物的回收率為83.4%~106%,測定值的RSD為1.9%~4.9%,滿足脂溶性維生素的實際檢測要求。
2.8 樣品分析
按照試驗方法分析一款顆粒型保健食品,該保健食品包含維生素A乙酸酯、維生素D3、DL-α-維生素E乙酸酯、維生素K1和維生素K2,標簽值分別為4.0,0.2,50.0,0.5,1.8 μg·g-1。結果顯示,上述物質的檢出量分別為4.83,0.225,58.6,0.593,2.05 μg·g-1,檢出量和標簽值基本一致,略高于標簽值。
本工作采用酶解法降解復雜基質保健食品中的脂肪和蛋白質,正己烷提取7種脂溶性維生素營養補充劑,超高效液相色譜-串聯質譜法測定其含量。和傳統皂化法比較,方法操作簡單、檢測快速、有機試劑消耗量少、環境友好度高,具有一定的推廣價值。
