基于溫度迭代校正自吸收效應的激光誘導擊穿光譜定量分析方法*
2024-04-01 08:01:04侯佳佳張大成馮中琦朱江峰
物理學報 2024年5期
關鍵詞:測量
侯佳佳 張大成 馮中琦 朱江峰
(西安電子科技大學光電工程學院,西安 710071)
激光誘導擊穿光譜(laser-induced breakdown spectroscopy,LIBS)是一種理想的實時在線檢測合金中微量元素的方法.然而在激光誘導擊穿產生的高密度等離子體中,自吸收通常是一種不期望出現的效應,它降低了譜線的真實強度,使譜線強度隨目標物質含量增長呈非線性,從而嚴重影響對目標中元素含量測量的準確性.本文提出了一種基于溫度迭代校正自吸收效應的方法,借助等離子體熱平衡輻射模型,對等離子體電子溫度(T)和輻射粒子數密度乘以吸收路徑長度(Nl)這兩個參數進行迭代計算和校正,消除自吸收對譜線強度的影響,最終提高定量分析的準確性.對合金鋼樣品中Mn 元素的實驗測量結果表明,該方法有效地提高了Boltzmann 平面圖的線性度及元素含量的測量精度.該方法模型簡單,計算效率高,且與Stark 展寬系數的可用性和準確性無關,可以直接獲得輻射粒子數密度和吸收路徑長度參數,因此在提高LIBS 定量分析能力的同時,還可以實現對等離子體狀態的診斷.
1 引言
由于激光誘導擊穿光譜(laser-induced breakdown spectroscopy,LIBS)技術具有分析速度快、多元素同時檢測、無需樣品制備、可原位和非接觸測量等優點,因此已被作為一種理想的物質元素實時在線檢測方法[1-7].然而,在LIBS 產生的高密度等離子體中存在自吸收效應,該效應不僅降低了被測元素譜線的真實強度,增加了其譜線寬度,也會影響等離子體的表征參數,從而影響最終定量分析的準確度和檢測限(limit of detection,LOD)[8,9].為了削弱自吸收的不利影響,國內外提出了許多自吸收的校正和消除方法,常見的有生長曲線(curve of growth,COG)法、自吸收系數校正法、等離子體建模法、內標參考線校正法、激光/微波輔助激發法等.例如,Aguilera等[10]使用COG 方法探索了等離子體的空間不均勻性和時間演化特性,并在等離子體的各個空間區域和演化階段消除了不同程度的自吸收引起的定標曲線的飽和.Mansour[11]通過分析譜線與光學薄Hα線的電子數密度比,校正了鋁原子譜線的自吸收效應,將電子溫度值 從1.407—1.255 eV 校正至 更準確 的1.283—0.896 eV.Gornushkin[12]建立了光學厚非均勻等離子體的理論模型,通過考慮連續輻射、吸收過程和吸收系數來確定譜線的自吸收程度.Sun 和Yu[13]采用內標參考線校正了自由定標 LIBS(CFLIBS)中的自吸收效應,提高了Boltzmann 平面圖的線性度和定量分析的準確性.Li等[14]和Tang等[15]提出了激光輔助激發LIBS (LSA-LIBS)和微波輔助激發LIBS (MA-LIBS)技術,通過使用連續波長可調諧激光器和連續微波發生器重新激發等離子體以降低其基態原子數密度,使Mn,Na,K 和Al 元素輻射線的半高全寬(full width at half maximum,FWHM)分別減少了25%,43%,58%和52%.Li等[16]提出了黑體輻射參考的自吸收校正方法(BRR-SAC)對CF-LIBS 中的自吸收進行了校正,將兩個樣品的平均測量誤差分別從3.62%和2.08%降低到0.27%和0.21%.Zhang等[17]提出了以等離子體圖像為關鍵參數的“相對自吸收系數”來評估自吸收,通過校正譜線強度獲得了高線性的定標曲線.上述自吸收校正方法都在一定程度上提高了LIBS 的定量分析性能,具有各自的特殊優勢和應用領域.然而,由于激光與被測物質相互作用的復雜性、等離子體演化的快速性和不均勻性以及環境和采集條件的差異性,使得等離子體中的自吸收效應隨時間和空間而復雜變化.
為了進一步全面地認識自吸收效應,本文從理論上建立了自吸收與相應輻射譜線參數、輻射躍遷類型、等離子體特征參數之間的定量關系模型,并通過研究電子溫度對自吸收的影響,提出了一種基于溫度迭代校正自吸收效應的方法,提高了LIBS定量分析的準確度.
2 實驗樣品及裝置
微量元素的種類和含量對于合金的性能和應用領域有著重要的影響[18-20].例如,錳元素(Mn)可以提高合金鋼的強度、硬度、彈性、淬透性和熱加工性,減弱硫元素的不利影響,但錳含量過高又會影響合金鋼的焊接性和耐蝕性.一般認為,當錳鋼中Mn 含量小于1.0%時,能提高鋼的強度和韌性,當Mn 含量為2.5%—3.5%時,相應的低錳鋼極易脆化,當Mn 含量大于13%時,制得的高錳鋼則既堅硬又富有韌性.隨著合金的廣泛應用,LIBS對于合金中微量元素的精確檢測,對于指導合金的生產和應用具有重要意義.因此,本工作中以6 個中低合金鋼標準樣品(國標號:JZG201—206)為測量樣品,且樣品表面已進行拋光處理.各樣品中微量元素Mn 的含量及不確定度如表1 所列.
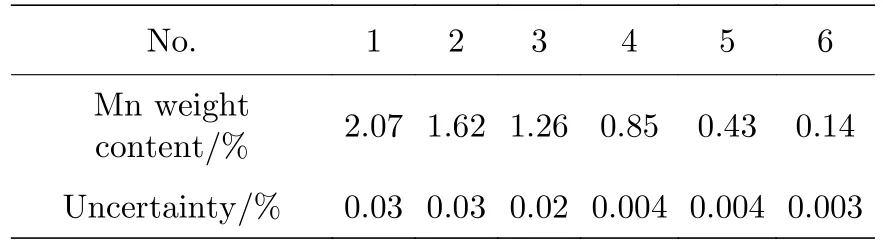
表1 中低合金鋼標準樣品中微量元素Mn 的質量含量及不確定度Table 1.Certified weight contents and uncertainty of minor element Mn in the middle-low alloy steels.
本實驗中使用了課題組自研的LIBS 裝置進行合金的光譜測量,其結構在之前的工作中已有詳細描述[21].采用1064 nm 波長的Nd:YAG 脈沖激光器(Dawa-300,Beamtech,CHN)作為等離子體燒蝕光源,其脈沖寬度為7 ns,重復頻率為10 Hz,單脈沖能量為30 mJ.激光束通過60 mm 焦距的石英透鏡聚焦于樣品表面下方0.5 mm 處以避免空氣擊穿,最大限度地減少空氣對發射光譜的干擾,獲得相對穩定的等離子體.樣品表面聚焦光斑直徑約為600 μm.在垂直于激光入射的方向,用一個焦距為50 mm 的石英透鏡采集等離子體輻射信號并通過200 μm 芯徑的光纖傳送到商用六通道光譜儀(AvaSpec Multi-channel,Avantes,NLD),其光譜覆蓋范圍為220—880 nm,分辨率為0.08—0.11 nm,積分時間為2 ms.激光器和光譜儀由數字延遲發生器(DG645,SRS Inc.,USA)觸發.為了減少等離子體連續輻射背景的干擾,將光譜采集時間相對于激光脈沖的延遲優化為500 ns.
3 溫度迭代校正自吸收的原理
假設等離子體是均勻的,并且在光譜采集期間處于局部熱力學平衡(LTE)條件下,則自吸收系數SA 可以通過實際測量到的發譜線的峰值強度I(λ0)與預期的無自吸收譜線峰值強度I0(λ0)(將光學薄條件下的COG 曲線有效地外推到與實際測量的輻射粒子相同數密度時而獲得的譜線強度)的比值表示,即[22]
其中K是代表吸收程度的參數;Δλ0是光學薄等離子體發射譜線的固有半寬.由(1)式可知,SA 隨著K參數的增加而減小,即K越大,發射譜線的自吸收越嚴重.
通過結合Boltzmann 分布律和反映振子強度與躍遷概率關系的Ladenburg 公式,K參數可以表示為[15]
其中N是原子或離子態的輻射粒子總密度;l是光傳播的吸收路徑長度;Aki是躍遷概率;gk是上能級簡并度;Z(T)是配分函數;Ei是下能級能量;kB是玻爾茲曼常數;T是等離子體電子溫度.由上述方程可知,自吸收與譜線的躍遷幾率、上能級簡并度、中心波長,等離子體的粒子數密度及吸收路徑長度成正相關,與譜線的下能級能量成負相關.值得注意的是,自吸收和溫度之間的關系隨譜線躍遷下能級而變化.
由(3)式可知,當所有的Ei-Ej都大于0 時,即Ei很大時,T越大,自吸收越嚴重;當Ei-Ej均小于0 時,即Ei很小時,T越大,自吸收越弱.換言之,當譜線躍遷的下能級很低或者處于基態時,自吸收隨等離子體溫度的升高而減小;反之,當譜線躍遷的下能級處于相對較高的激發態,則自吸收隨等離子體溫度的升高而增大.
由以上理論分析可知,等離子體電子溫度T、輻射粒子的數密度乘以吸收路徑長度參數Nl可以決定自吸收的程度.考慮到Boltzmann 平面圖描述了譜線強度與等離子體電子溫度之間的關系,而光學薄條件下理想的電子溫度T和參數Nl不僅可以使Boltzmann 平面圖具有最佳的線性,并且會符合Boltzmann 平面圖對等離子體電子溫度的表征.由此可以得出一種基于溫度迭代校正自吸收的方法,其原理如下:對于LIBS 實驗中獲得的譜線,可以根據Boltzmann 平面圖得到對應分析元素的初始等離子體電子溫度T0,設定一個初始輻射粒子數密度和吸收路徑長度參數Nl0后,則通過等離子體溫度T0可以計算出譜線的自吸收系數SA0并對譜線強度進行一次校正.然后,根據校正后的譜線強度得到新的Boltzmann 平面圖并評估及線性度,通過調整Nl參數使其線性度達到最優,之后對比此時得到的溫度與初始溫度是否一致,如果有差異則采用此時的溫度及調整后的Nl值再次得到譜線的自吸收程度,對譜線進行二次校正.如此迭代循環,直到得到的電子溫度收斂,則表明自吸收校正完畢.其具體步驟如流程圖1 所示[23].

圖1 基于溫度迭代校正自吸收方法的流程圖Fig.1.Flowchart of the self-absorption correction method based on temperature iteration.
至此可以獲得準確的分析譜線強度、等離子體電子溫度、輻射粒子數密度及吸收路徑長度,并且根據自吸收和譜線半寬的關系,可以進一步得到準確的光學薄譜線固有展寬.應當注意的是,本工作中使用的譜線強度是積分強度,自吸收對測得的譜線積分強度(λ)的影響可以根據SA 系數和無自吸收的積分強度(λ)進行數值計算如下[22]:
其中β=0.46.
4 結果與討論
圖2 是Mn 重量含量為2.07%的1 號合金鋼標樣在380—670 nm 范圍內的典型光譜.其中Mn I 383.44,403.31,404.14,475.40,476.23,478.34 和482.35 nm 七條譜線較強且穩定性較好,用于構建Boltzmann 平面圖以獲得等離子體電子溫度T.具有光學薄特性的Hα線(656.27 nm)可用于測定等離子體的電子密度ne,通過結合Mn I 476.23 nm譜線的半寬,可以按照自吸收系數校正法[22]計算自吸收系數,并與本文提出的溫度迭代校正法計算的自吸收系數進行比較.圖2 中所附的放大圖為波長從381 到405 nm 的細節圖,更清晰的展示出了Mn I 383.44,403.08,403.31,403.45 和404.14 nm等譜線.
表2 詳細列出了來自NIST 數據庫的Mn I 譜線等光譜參數.Fe I 400.52 nm 和Fe I 489.15 nm譜線分別用于Mn I 383.44,403.31,404.14 nm 三條譜線和Mn I 475.40,476.23,478.34,482.35 nm四條譜線的內標歸一化.為了提高等離子體發射光譜測量的重復性,補償樣品的不均勻性,本實驗采集了在相同的實驗條件下樣品表面100 個不同位置的300 幅光譜,并對其進行了扣除連續背景和數據平均處理.
4.1 溫度迭代校正自吸收LIBS 的定量分析
圖3 為500 ns 延遲下利用Mn I 476.23 nm譜線對幾種標樣中Mn 含量進行分析的定標曲線,其中圖3(a)為使用原始譜線強度直接定標的結果,圖3(b)為使用與Mn I 476.23 nm 線相鄰的內標Fe I 489.15 nm 線進行內標歸一化后的強度定標的結果,且圖中以誤差棒的形式表明了所用譜線強度的測量不確定度.其中譜線強度的測量不確定度是以5 個點的采樣數據為一組,將300 幅光譜分為20 組,分析獲得組間平均后譜線強度的相對標準偏差來表征.
從圖3(a)可以看出,由于樣品中元素分布的不均勻性、激光能量和等離子體羽流的波動等原因導致輻射譜線強度波動較大,其平均不確定度為11.66%,這些影響因素導致使用原始譜線強度的定標曲線的線性度非常差.而在圖3(b)中,使用內標歸一化強度的定標曲線的線性度大大提高到0.971,且其譜線強度的平均不確定度降低至6.31%,表明內標法可以有效降低譜線強度的波動,但由于Mn 線和內標Fe 線各自的自吸收影響,線性度仍需進一步提高.為了評估無自吸收校正和有自吸收校正時LIBS 的測量準確性,在實驗中選擇Mn 質量含量為0.14%,0.43%,0.85%,1.26%和2.07%的合金作為定標樣品,選擇Mn 質量含量為1.62%的合金作為預測樣品(標記為空心點).結果表明,只使用內標歸一化而不進行自吸收校正的情況下,預測樣品中Mn 的分析質量含量為1.55%,其測量相對誤差(RE)為4.32%.
根據提出的基于溫度迭代校正自吸收的方法,對選定的7 條分析Mn 線和相應的內標Fe 線進行了自吸收校正.圖4 給出了Mn 質量含量為2.07%合金鋼樣品在自吸收校正前后的Boltzmann 平面圖,可以看出,在自吸收校正前各點非常分散,線性相關系數R2為0.867.而自吸收校正后,這些點與相應的直線擬合線重合地更好,線性相關系數R2提高到了0.974.結果表明,在校正自吸收效應后,可以有效提高Boltzmann 平面圖的線性度,且得到的等離子體電子溫度由1.13 eV 校正為更準確的0.91 eV.

圖4 Mn 質量含量為2.07%的合金樣品自吸收校正前后Boltzmann 比較圖Fig.4.Comparison of Boltzmann plots that without and with self-absorption (SA) correction for the 2.07% Mn alloy sample.
圖5 顯示了對Mn I 476.23 nm 譜線及內標Fe 線進行自吸收校正后再進行內標歸一化后的定標曲線,以及預測樣品的定量分析結果(標記為空心點).將分析Mn 線的歸一化線強度代入相應的線性校正方程,得到自吸收校正后預測樣品中的Mn 質量含量為1.6%,其測量相對誤差為1.23%.另外,經過自吸收校正后的內標歸一化譜線強度的不確定度可以進一步降低至4.41%,這主要是因為每次測量時激光能量的波動及等離子體演化的快速性和不均勻性導致等離子體中的自吸收效應隨時間和空間而復雜變化,因此譜線自吸收程度的差異也導致了譜線強度的波動,所以經過自吸收校正后可以進一步降低譜線強度的不確定度.
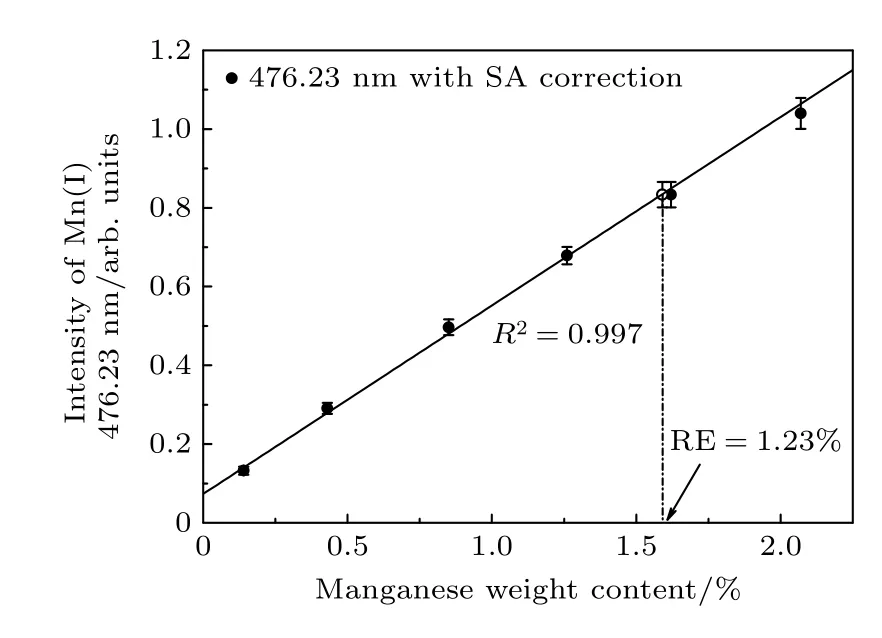
圖5 Mn I 476.23 nm 譜線經過自吸收校正后的內標歸一化定標曲線Fig.5.Calibration curves of Mn I 476.23 nm using the internal standard normalized intensity with self-absorption correction.
為了體現出該溫度迭代校正自吸收方法對LIBS 定量分析結果的影響,表3 列出了對所有樣品即不同Mn 元素質量含量的測量相對誤差及平均誤差.

表3 中低合金鋼標準樣品中微量元素Mn 質量含量的測量相對誤差Table 3.Measurement relative error of minor element Mn in the middle-low alloy steels.
由表3 可知,經過自吸收校正后對樣品中Mn元素質量含量的測量相對誤差均有所提高,其平均測量相對誤差由自吸收校正前的10.99%提升至校正后的4.93%.上述定量分析結果表明,LIBS 在內標歸一化前對分析譜線和內標譜線進行基于溫度迭代的自吸收校正,可有效地提高定標曲線的線性度和定量分析的準確度.
4.2 溫度迭代校正自吸收LIBS 的驗證
為了驗證自吸收校正的效果,此處使用經典自吸收系數法對SA 進行了計算,并與本文提出的基于溫度迭代校正方法得到的系數SA 進行了比較.
對于Stark 展寬系數已知的譜線,自吸收系數SA 可以表示為[22]
其中α=-0.54;Δλ 是測量譜線的半寬;ws是Stark展寬系數;ne是光學薄等離子體中的電子密度.對于金屬合金的分析,通常可以根據如下關系式從Hα線的半寬得到此電子密度:
其中ΔλH是Hα線的固有半高全寬;α1/2是Hα線簡化Stark 線型的半高全寬(單位:?),一個弱依賴于電子密度和溫度的系數,Balmer 線系的精確α1/2值可在文獻[24]中找到.
圖6 中用紅色柱顯示了由(5)式計算的Mn I 476.23 nm 譜線的自吸收系數,藍色柱顯示了用溫度迭代校正方法計算的自吸收系數.從圖6 中可以看出,經典自吸收系數法得到的SA 與本文提出的校正方法得到的SA 基本一致,說明溫度迭代法可以有效地校正譜線的自吸收.兩種方法計算的自吸收系數之間的最大偏差為7.1%,我們認為這種偏差是由于在計算SA 時使用的Stark 展寬系數ws的不準確造成的,本文使用的ws是在T=10000 K,ne=1016cm-3條件下的測量值[25].另外,由使用分析Mn I 線原始強度直接定標的結果可知,由于樣品的不均勻性等原因導致激發的等離子體中元素含量與標稱含量不一致,且由于每個樣品激發的等離子體的溫度有一定的差別,所以導致本實驗中分析Mn I 線的自吸收程度不隨著Mn 元素含量的增大而增大.
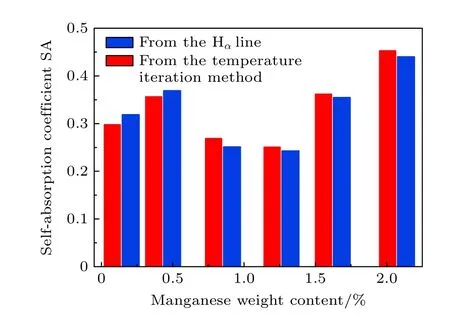
圖6 Mn I 476.23 nm 譜線的自吸收系數SAFig.6.The self-absorption coefficients (SA) for Mn I 476.23 nm line.
4.3 分析與討論
從以上對合金鋼中Mn 元素含量的分析過程可以看出,本文提出的基于溫度迭代校正自吸收的方法具有物理原理清晰、計算效率高、無需譜線Stark 展寬系數等優點.它能有效地提高定量分析的準確度,并獲得準確的等離子體電子溫度、輻射粒子數密度和吸收路徑長度參數.
該自吸收校正方法的局限性主要體現在以下方面.因為等離子體參數隨時間和空間的變化而變化,所以使用Boltzmann 平面圖計算電子溫度時需要等離子體處于局部熱平衡近似條件,由于該條件在等離子體產生和膨脹的最初時期(等離子體膨脹特征時間大于碰撞平衡時間)及等離子體壽命的最后時期(等離子體冷卻且電子數密度低于McWhirter 準則給出的極限值)不成立,這就要求光譜信息需要在有限的時間窗口采集才能成立.然而,時間分辨寬帶光譜儀由于其價格昂貴、體積較大、以及對外部條件的敏感性,通常不適合用于現場及便攜式LIBS 儀器.只帶有CCD 的寬帶光纖光譜儀雖然只能提供時間積分LIBS 光譜,但由于其價格低廉,性能穩定可靠,因此對于現場及便攜式LIBS 儀器的使用具有更實用的價值.事實上,由于LIBS 光譜強度是采集延遲時間的指數衰減函數(見圖7),等離子體輻射的主要強度在初始時期,時間積分測量的主要信號也來于自等離子早期.因此有理由假設在一個給定延時下的時間積分光譜的主要貢獻將來自于非常有限的一個與信號衰減時間同數量級的時間窗口,在該等效時間窗口內,等離子體滿足局部熱平衡近似,等離子體電子溫度及電子密度等參數可以等效為一個平均值,從而可以模擬時間分辨測量的效果.Grifoni等[26]對比了時間積分LIBS 光譜和時間分辨LIBS 光譜,結果表明由時間分辨光譜評估得到的等離子體電子溫度與電子密度與時間積分光譜得到的參數基本相同.并且從本實驗的實際效果來看,通過校正等離體平均溫度能夠有效提高測量結果的準確性,也表明了時間積分測量的結果主要來自等離子體早期的光譜強度.
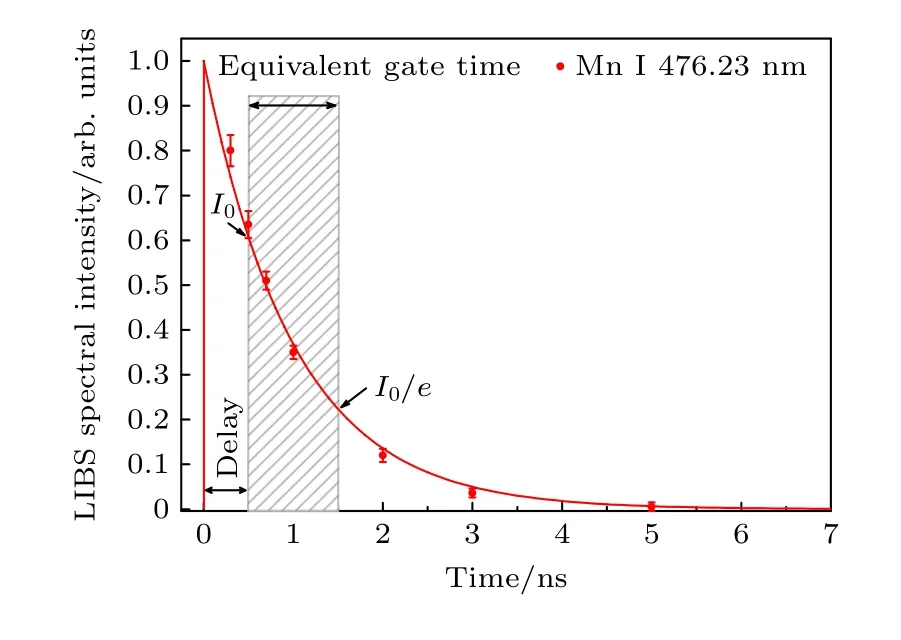
圖7 時間積分LIBS 分析中的等效門時間Fig.7.Equivalent gate time in time-integrated LIBS analysis.
實驗中使用的譜線強度為積分強度,但由于分析的Mn 元素在合金中的質量含量只有0.14%—2.07%,譜線強度本身不夠強,且合金中譜線過于密集,部分所選的分析譜線不夠獨立,受到了其他譜線的影響,這都導致譜線積分強度有一定的誤差.且雖然在等效時間窗口內可以認為等離子體滿足局部熱平衡近似,但是時間積分光譜與理想的物理假設還是存在一定的偏差.因此,通過優化實驗條件和樣品處理,更換分辨率更高的光譜儀以減小譜線的干擾,采用時間分辨光譜并結合對譜線線型的擬合,在盡量消除其他因素對譜線強度測量影響的前提下,定量分析結果還可以進一步提升.
5 結論
本文提出了一種基于溫度迭代校正自吸收的方法,通過連續計算和校正等離子體電子溫度T和輻射粒子數密度乘以吸收路徑長度參數Nl來校正自吸收效應.合金鋼樣品的實驗結果表明,自吸收校正后,Boltzmann 平面圖的線性度由未經自吸收校正的0.867 增加到0.974,Mn 元素的單變量定標曲線的線性相關系數R2由0.971 提升到0.997,對Mn 質量含量為1.62%的測試合金鋼樣品的元素測量相對誤差由自吸收校正前后的4.32%提升為1.23%.此外,經典自吸收系數法與本文溫度迭代校正法得到的SA 系數的一致性表明了該方法對自吸收校正的有效性.由于該方法不依賴于Stark 展寬系數的可用性和準確性,且可以獲得準確的等離子體電子溫度、輻射粒子數密度和吸收路徑長度參數,將有利于等離子體診斷和定量分析.
猜你喜歡
小學科學(學生版)(2021年5期)2021-07-22 02:40:06
中學生數理化·八年級物理人教版(2019年9期)2019-11-25 07:33:02
中學生數理化·八年級物理人教版(2019年3期)2019-04-25 06:20:54
中學生數理化·八年級物理人教版(2018年3期)2018-05-31 08:52:45
數學小靈通(1-2年級)(2017年10期)2017-11-08 08:39:45
軍事文摘·科學少年(2017年4期)2017-06-20 23:25:16
軍事文摘·科學少年(2017年2期)2017-04-26 21:58:43
中學生數理化·八年級物理人教版(2016年3期)2016-04-07 04:49:32
少兒科學周刊·兒童版(2016年1期)2016-03-14 03:52:21
閱讀與作文(小學低年級版)(2015年4期)2015-04-29 00:00:00
