TYK2抑制劑BMS986202的合成工藝
2024-04-27 09:40:06張倩倩李銘東黨詩涵李庶心胡文祥
合成化學(xué) 2024年4期
關(guān)鍵詞:體系
張倩倩, 李銘東, 黨詩涵,, 李 春,,, 李庶心, 胡文祥,4*, 閔 清*
(1. 湖北科技學(xué)院 藥學(xué)院,湖北 咸寧 437100; 2. 江西中醫(yī)藥大學(xué) 藥學(xué)院,江西 南昌 330000; 3. 蘇州隆博泰藥業(yè)有限公司,江蘇 蘇州 215000; 4. 北京神劍天軍醫(yī)學(xué)科學(xué)研究院 京東祥鵠微波化學(xué)聯(lián)合實驗室,北京 101601)
Deucravacitinib(BMS986165)是全球首款上市的口服選擇性酪氨酸激酶2(TYK2)變構(gòu)抑制劑,也是首款原創(chuàng)性氘代新藥。BMS986165由百時美施貴寶(BMS)公司研發(fā),化學(xué)結(jié)構(gòu)見圖1,并于2022年9月9日被美國食品和藥物管理局批準(zhǔn)用于適合全身治療或光療的中重度斑塊型銀屑病成人患者[1]。 TYK2也是JAK家族,包括JAK1, JAK2, JAK3和TYK2中的一員。JAK抑制劑在炎癥和癌癥等治療領(lǐng)域取得了很大成功,但它們在臨床應(yīng)用中容易引起嚴(yán)重感染、惡性腫瘤和血栓等嚴(yán)重副作用[2-4]。由于調(diào)控的細胞信號通路不同,相比于副作用嚴(yán)重的JAK抑制劑,高選擇性的TYK2在治療多種自身免疫性疾病,例如斑塊型銀屑病[5]、銀屑病關(guān)節(jié)炎[6]、系統(tǒng)性紅斑狼瘡[7]、炎性腸病[8]、類風(fēng)濕性關(guān)節(jié)炎[9]和斑禿[10]等的同時,避免了泛抑制帶來的副作用[11]。 BMS986165選擇性結(jié)合TYK2假激酶(JH2)結(jié)構(gòu)域,并通過穩(wěn)定調(diào)節(jié)JH2結(jié)構(gòu)域來阻斷受體介導(dǎo)的TYK2激活,從而抑制白細胞介素IL-23、 IL-12和干擾素IFN的信號傳導(dǎo)[12]。 BMS986165選擇性與TYK2的調(diào)控域結(jié)合,實現(xiàn)對TYK2及其下游信號的變構(gòu)抑制,相對其他JAK亞型具有較高的選擇性,在建議的治療劑量下,不會抑制JAK1, JAK2和JAK3的活性[13]。

圖1 BMS986165的結(jié)構(gòu)
BMS986202(圖2)是百時美施貴寶公司基于BMS986165發(fā)現(xiàn)的一種新的臨床候選藥物。研究人員基于結(jié)構(gòu)優(yōu)化,將BMS986165骨架中的噠嗪優(yōu)化為吡啶,再對苯環(huán)取代基雜環(huán)的對位進行取代,以占據(jù)朝向Thr599殘基的疏水空腔,由此發(fā)現(xiàn)了BMS986202。 BMS986202具有較好的活性、水溶性和滲透性。經(jīng)過進一步的藥代性質(zhì)評價,研究人員發(fā)現(xiàn)其具有較優(yōu)秀的藥動學(xué)性質(zhì),口服生物利用度極好。同時,研究人員通過IL-12/IL-18誘導(dǎo)小鼠干擾素產(chǎn)生的體內(nèi)模型、IL-23誘導(dǎo)的皮膚炎癥模型和結(jié)腸炎模型,系統(tǒng)評價了化合物BMS986202的體內(nèi)活性,發(fā)現(xiàn)其均表現(xiàn)出顯著的體內(nèi)活性[14]。目前,BMS986202正處于Ⅱ期臨床試驗,具有良好的開發(fā)和應(yīng)用前景。

圖2 BMS986202的結(jié)構(gòu)
目前,專利[15]報道的BMS986202合成方法主要是以2-溴-6-硝基苯酚為起始原料,經(jīng)取代、還原、偶聯(lián)和芳基化,最后再與環(huán)丙甲酰胺進行酰胺化反應(yīng)制得。步驟較少,收率較高,但在中間體的純化上,需要通過柱層析來完成,存在反應(yīng)周期長,負載量低,時間成本增加的問題,不適合工業(yè)化生產(chǎn)。
本研究參考原研路線(圖3),對BMS986202的合成工藝進行了優(yōu)化,產(chǎn)品結(jié)構(gòu)經(jīng)1H NMR分析表征。制備中間體2時,原研專利[15]所用溶劑為乙醇和水,反應(yīng)溫度為室溫,通過柱色譜純化,收率為85%。本研究以甲醇作為溶劑,控制反應(yīng)溫度約20 ℃,反應(yīng)達到完全且雜質(zhì)少,無需純化可直接用于后續(xù)反應(yīng),避免了原研專利通過快速柱色譜純化產(chǎn)物,大幅度地簡化了操作,最終收率為81%。制備中間體4時,原研專利[15]的合成路線以6-溴-2-硝基苯酚為起始原料,經(jīng)取代、還原和偶聯(lián)3步反應(yīng)制得(圖4),收率為60%。然而3步反應(yīng)步驟繁瑣,中間體均需要通過柱層析進行純化,存在操作復(fù)雜、耗時長和成本增加的問題,不適合放大生產(chǎn)。因此,本研究直接將1-溴-2-甲氧基-3-硝基苯作為原料,既簡化了步驟,又節(jié)省了時間和成本。起初,以1-溴-2-甲氧基-3-硝基苯為原料,想先通過偶聯(lián)反應(yīng)再還原制得中間體4(圖5)。然而,在實驗過程中,本研究發(fā)現(xiàn)大約有75%的原料未反應(yīng)完全,收率很低。因此,最終選擇先還原再偶聯(lián)的方法,最后收率為72%。在制備中間體6時,原研專利[15]采用的鈀試劑為[1,1′-二(二叔丁基膦)二茂鐵]合二氯鈀(II),堿為磷酸氫二鉀,反應(yīng)溫度為100 ℃,收率較低,為75%。隨后,本實驗將鈀試劑改為1,1′-雙二苯基膦二茂鐵二氯化鈀,堿改為碳酸鈉,反應(yīng)溫度降至75 ℃,體系干凈,雜質(zhì)少,后處理簡單,收率得到提高,為81%。制備中間體7時,將氯代試劑改為氯化亞砜,堿改為三乙胺,反應(yīng)時間縮短,純度提高,后處理簡單,收率由原研專利[15]的74%提高到81%。而重復(fù)專利[15]制得中間體7,反應(yīng)時間長,雜質(zhì)多,需要通過柱層析進行純化,操作復(fù)雜且繁瑣。制備中間體8時,本研究使用四氫呋喃為反應(yīng)溶劑,沸點低,避免專利[15]中處理N,N-二甲基乙酰胺的步驟,后處理更加簡單,方便,收率由79%提高至84%。制備目標(biāo)化合物BMS986202時,本研究將原研專利[15]的溶劑由二氧六環(huán)改為1,4-二氧六環(huán)和水的混合溶劑,反應(yīng)溫度由130 ℃降至90 ℃,反應(yīng)收率由原研專利的70%提高到85%。

圖3 BMS986202的合成路線[15]

圖4 中間體4的合成路線[15]

圖5 本文中間體4的合成路線
1 實驗部分
1.1 儀器與試劑
ZF-20C型暗箱式自動紫外分析儀; RY-1G型熔點儀(溫度未校正); Varian 400 Hz型核磁共振儀(CDCl3或DMSO-d6為溶劑,TMS為內(nèi)標(biāo))。
化合物1,成都伊諾達博醫(yī)藥科技有限公司;化合物3,畢德醫(yī)藥有限公司;化合物5,江蘇艾康生物醫(yī)藥研發(fā)有限公司;化合物9,薩恩化學(xué)技術(shù)(上海)有限公司;甲醇、叔丁醇、1,4-二氧六環(huán),江蘇強盛功能化學(xué)有限公司;四氫呋喃,無錫市晶科化工有限公司。所用試劑均為分析純。
1.2 合成
(1) 3-溴-2-甲氧基苯胺(中間體2)的合成
將原料1-溴-2-甲氧基-3-硝基苯(30.0 g, 129.3 mmol)加入甲醇溶液(80 mL)中,再加入氯化銨(45.0 g, 841.3 mmol)。將其置于冰水浴中攪拌10 min,然后緩慢加入鋅粉(67.2 g, 103.4 mmol),速度以控制體系溫度不超過20 ℃為宜,加畢,繼續(xù)冰水浴下(0 ℃)反應(yīng)2 h(TLC檢測)。體系過硅藻土抽濾,濾液減壓濃縮,加入飽和碳酸鈉水溶液(150 mL),然后用乙酸乙酯(3×50 mL)萃取,合并有機相,再用飽和氯化鈉水溶液(50 mL)洗滌,經(jīng)無水Na2SO4干燥,蒸除溶劑,得到粗品中間體221.2g,收率81%,直接投入下一步反應(yīng)。
(2) 2-甲氨基-3-(4,4,5,5-四甲基-1,3,2-二氧雜硼烷-2-基)苯胺(中間體4)的合成
將原料3-溴-2-甲氧基苯胺(21.2 g, 90.6 mmol)和聯(lián)硼酸頻那醇酯(27.6 g, 108.7 mmol)依次加入叔丁醇(80 mL)溶液中(20 ℃),電磁攪拌,然后加入2-乙基己酸鉀(49.5 g, 271.7 mmol),抽真空,N2置換,加入PdCl2(dppf)(0.7 g, 0.9 mmol),再次抽真空,N2置換3次,然后加熱,升溫至80 ℃回流反應(yīng)4 h(TLC檢測)。體系過硅藻土抽濾,濾液減壓濃縮后加入冰水(150 mL),然后用乙酸乙酯(3×50mL)萃取,合并有機相,再用飽和氯化鈉水溶液(50 mL)洗滌,經(jīng)無水Na2SO4干燥,蒸除溶劑,殘余物經(jīng)石油醚打漿純化得中間體4,棕色固體,16.3 g, m.p.101~103 ℃,收率72%;1H NMR(400 MHz, CDCl3)δ:7.14~7.11(m, 1H), 6.93(t,J=15.2 Hz, 7.6 Hz, 1H), 6.87~6.85(m, 1H), 3.83(3, 3H), 1.36(s, 12H)。
(3) 3-(5-氟嘧啶-2-基)-2-甲氧基苯胺(中間體6)的合成
將碳酸鈉(17.4 g, 163.8 mmol)加入250 mL三口燒瓶中,加水(60 mL)攪拌,然后加入原料2-甲氨基-3-(4,4,5,5-四甲基-1,3,2-二氧雜硼烷-2-基)苯胺(16.3 g, 65.5 mmol)和2-溴-5-氟嘧啶(12.7 g, 72.1 mmol),再滴加1,4-二氧六環(huán)(70 mL),電磁攪拌,抽真空,N2置換,加入PdCl2(dppf)(2.7 g, 3.7 mmol),再次抽真空,N2置換3次,加熱,升溫至75 ℃反應(yīng)4 h(TLC檢測)。體系過硅藻土抽濾,濾液減壓濃縮,加入冰水(150 mL),用乙酸乙酯(3×50mL)萃取,合并有機相,再用飽和氯化鈉水溶液(50 mL)洗滌,經(jīng)無水Na2SO4干燥,蒸除溶劑,得中間體6,黃色油狀液體,11.6 g,收率81%,粗品,直接投入下一步反應(yīng)。1H NMR(400 MHz, DMSO-d6)δ:8.96~8.94(d,J=0.8 Hz, 2H), 7.23~7.20(m, 1H), 7.15~7.12(m, 1H), 6.95~6.90(m, 1H), 5.25(s, 2H), 3.83(s, 3H)。
(4) 4,6-二氯吡啶-氘甲基酰胺(中間體7)的合成
將原料4,6-二氯煙酸(20.0 g, 104.2 mmol)投入250 mL三口燒瓶中,然后加入氯化亞砜(50 mL),同時滴加催化劑量的N,N-二甲基甲酰胺,電磁攪拌。加熱,升溫至80 ℃回流反應(yīng)。體系逐漸澄清、透明,呈淡黃色。1.5 h后(TLC檢測),將體系濃縮,旋干。將殘留物溶于二氯甲烷中,再次濃縮,旋干,以確保完全除去多余的二氯亞砜,得到化合物4,6-二氯煙酰氯,直接用于下一步反應(yīng)。提前配置游離的氘代甲胺溶液:氘代甲氨鹽酸鹽(8.1 g, 114.6 mmol)加水,再加入三乙胺(11.6 g, 114.6 mmol),同時滴加二氯甲烷溶液(20 mL)。配置好后為透明混懸液,封口放入冰箱冷藏。將化合物4,6-二氯煙酰氯溶于二氯甲烷溶液(30 mL)中,然后將其置冰水浴冷卻至0 ℃,緩慢滴加配置好的氘代甲胺溶液,滴加速度以保持體系溫度不超過5 ℃為宜。滴加過程中,體系中有白色固體析出。滴加完畢,繼續(xù)冰水浴下反應(yīng)2.5 h(TLC檢測)。加冰水(150 mL)將體系淬滅,用二氯甲烷(3×50 mL)萃取,合并有機相,用飽和K2CO3水溶液(50 mL)洗滌,再用飽和氯化鈉水溶液(50 mL)洗滌,經(jīng)無水Na2SO4干燥,蒸除溶劑,殘余物經(jīng)石油醚打漿,純化,得到中間體7,白色固體,17.5 g,收率81%, m.p.135~137 ℃;1H NMR(400 MHz, DMSO-d6)δ:8.57(s, 1H), 8.45(s, 1H), 7.90(s, 1H)。
(5) 6-氯-4-((3-(5-氟嘧啶-2-基)-2-甲氧基苯基)氨基)-N-氘甲基煙酰胺(中間體8)的合成
將3-(5-氟嘧啶-2-基)-2-甲氧基苯胺(11.6 g, 58.2 mmol)和中間體7(12.1 g, 57.9 mmol)依次加入裝有四氫呋喃(30 mL)的250 mL三口燒瓶中,電磁攪拌;將其置于冰水浴中,待溫度降至0 ℃時,緩慢滴加LiHMDS(15 mL),滴加速度以控制體系溫度不超過20 ℃為宜,滴加完畢,撤去冰水浴,體系自然升至室溫(21 ℃)反應(yīng)3 h(TLC檢測)。體系由墨綠色逐漸變成棕色。體系加冰水(5 mL)淬滅,過硅藻土抽濾,濾液減壓濃縮,蒸除體系中的四氫呋喃,然后加入冰水(150 mL),用二氯甲烷(3×50 mL)萃取,合并有機相,再用飽和氯化鈉水溶液(50 mL)洗滌,經(jīng)無水Na2SO4干燥,蒸除溶劑,殘余物經(jīng)打漿(二氯甲烷 ∶石油醚=5 ∶1,V∶V)純化,過濾,收集濾餅,得中間體8,白色固體,18.2 g,收率84%, m.p.193~195 ℃;1H NMR(400 MHz, DMSO-d6)δ:10.31(s, 1H), 9.15(s,1H), 9.06~9.04(d,J=0.9 Hz, 2H), 7.95~7.90(s, 1H), 7.82~7.78(s, 1H), 7.77~7.75(m, 1H), 7.60~7.56(m, 1H), 7.46~7.41(m, 1H), 3.70(s, 3H)。
(6) 6-環(huán)丙烷甲酰胺基-4-((3-(5-氟嘧啶-2-基)-2-甲氧基苯基)氨基)-N-氘甲基煙酰胺的(BMS986202)合成
將碳酸銫(20.8 g, 63.8 mmol)投入250 mL三口燒瓶中,加水(30 mL)攪拌,然后加入原料6-氯-4-((3-(5-氟嘧啶-2-基)-2-甲氧基苯基)氨基)-N-氘甲基煙酰胺(10.0 g, 25.6 mmol),并滴加1,4-二氧六環(huán)(50 mL),抽真空,N2置換,加入Pd2(dba)3(1.2 g, 1.3 mmol)和Xantphos(1.5 g, 2.6 mmol),再次抽真空,N2置換3次,加熱,升溫至90 ℃反應(yīng)4.5 h(TLC檢測)。體系過硅藻土抽濾,濾液減壓濃縮,蒸除反應(yīng)溶劑后加入冰水(150 mL),用二氯甲烷(3×50 mL)萃取,合并有機相,再用飽和氯化鈉水溶液(50 mL)洗滌,經(jīng)無水Na2SO4干燥,蒸除溶劑,殘余物經(jīng)重結(jié)晶(二氯甲烷∶石油醚=3∶1,V∶V)純化,得產(chǎn)物BMS986202,白色固體,9.5 g,收率85%,純度98.8%, m.p.250~252 ℃;1H NMR(400 MHz, DMSO-d6)δ:10.73(s, 1H), 10.56(s, 1H), 9.05(d,J=0.9 Hz, 1H), 8.57(s, 1H), 8.52 (s, 1H), 8.14~7.95(m, 1H), 7.55(dd,J=7.8 Hz, 1.5 Hz, 1H), 7.43(dd,J=7.6 Hz, 1.3 Hz, 1H), 7.34~7.20(m, 1H), 3.65 (s, 3H), 2.08~1.84(m, 1H), 0.82~0.75(m, 4H); MS(ESI)m/z:calcd for C22H18D3FN6O3{[M+H]+}439.18, found 439.5。
2 結(jié)果與討論
2.1 中間體4的合成分析
在合成中間體4時,主要考察了鈀試劑、配體、堿和溶劑對Suzuki偶聯(lián)反應(yīng)的影響。結(jié)果發(fā)現(xiàn),該類反應(yīng)在大多數(shù)情況下,原料未能反應(yīng)完全。因此本研究嘗試了Pd2(dba)3和PdCl2(dppf) 2種鈀試劑,實驗結(jié)果顯示,使用Pd2(dba)3和配體Xantphos時,體系未反應(yīng)。本研究采用了無需配體參與的鈀試劑PdCl2(dppf),發(fā)現(xiàn)原料發(fā)生反應(yīng)。同時在以PdCl2(dppf)為鈀試劑的前提下,對該反應(yīng)的溶劑和堿進行了考察,最終確定了在反應(yīng)溫度為80 ℃條件下,以叔丁醇為溶劑,以2-乙基己酸鉀為堿是合成中間體4的最佳條件,收率為72%(表1, Entry 5)。
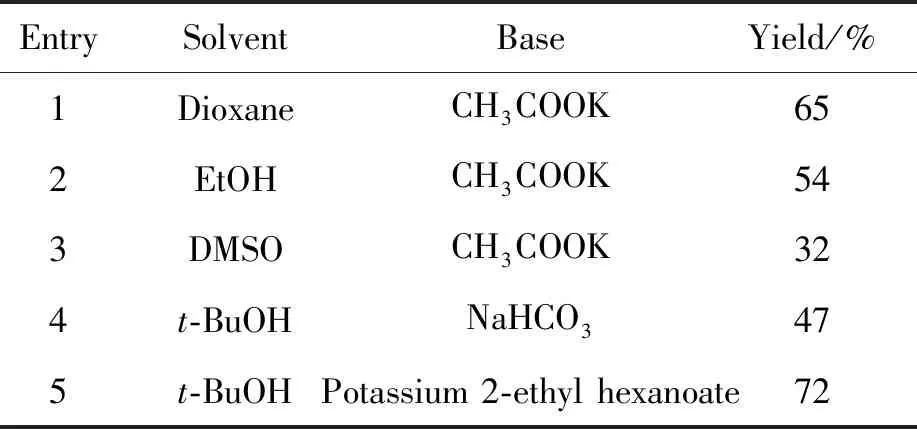
表1 溶劑、堿對中間體4收率的影響
2.2 中間體6的合成分析
類似中間體4,中間體6的合成過程同樣考察了鈀試劑、配體、堿、溶劑和溫度對Suzuki偶聯(lián)反應(yīng)的影響。
(1) 堿對Suzuki偶聯(lián)反應(yīng)的影響
從Suzuki偶聯(lián)反應(yīng)的機理可以看出,堿在整個反應(yīng)過程中起著重要的作用[16]。通常用于Suzuki偶聯(lián)反應(yīng)的堿既有有機堿也有無機堿。常用的無機堿有K3PO4、 K2CO3、 KOH、 Cs2CO3、 Na2CO3、 KF、 CsF和Ba(OH)2等;常用的有機堿有KOBu-t、 NaOBu-t、 KOMe、 NEt3和t-BuNH2等。本研究嘗試了Na2CO3, K3PO4和K2HPO43種堿對Suzuki偶聯(lián)反應(yīng)的影響,實驗結(jié)果顯示,K3PO4和K2HPO4均不能使原料反應(yīng)完全,收率較低。而用Na2CO3作為堿進行反應(yīng)時,原料反應(yīng)完全,體系雜質(zhì)少,后處理簡單,因此最終采用Na2CO3作為堿進行反應(yīng)(表2, Entries 1, 4)。
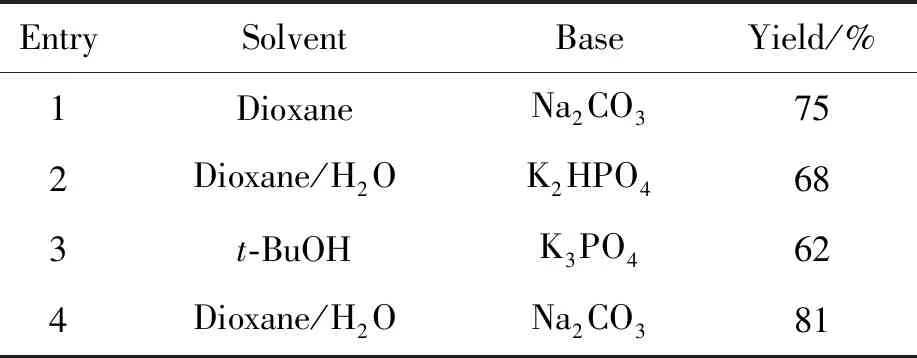
表2 溶劑、堿對中間體6收率的影響
(2) 反應(yīng)溶劑對Suzuki偶聯(lián)反應(yīng)的影響
同堿一樣,溶劑在Suzuki偶聯(lián)反應(yīng)中也起著重要的作用。它除了可以使參與反應(yīng)的各個組分處于均相之外,也是調(diào)節(jié)反應(yīng)溫度的載體。在Suzuki偶聯(lián)反應(yīng)中各類溶劑均有應(yīng)用,例如:DMF、二氧六環(huán)、THF、甲苯、二甲苯、乙腈、三氯甲烷、丙酮以及各種醇等。醇中包括甲醇、丙醇和丁醇等。此外,一些混合溶劑在該反應(yīng)中也有很好的表現(xiàn)。本研究嘗試了1,4-二氧六環(huán)、叔丁醇以及混合溶劑1,4-二氧六環(huán)/水3種溶劑對Suzuki偶聯(lián)反應(yīng)的影響,結(jié)果發(fā)現(xiàn),當(dāng)反應(yīng)溶劑為1,4-二氧六環(huán) ∶水=4 ∶1(體積比)時,原料反應(yīng)完全,體系干凈,收率較高,為81%(表2, Entry 4)。
(3) 反應(yīng)溫度對Suzuki偶聯(lián)反應(yīng)的影響
在本實驗過程中發(fā)現(xiàn),不同的反應(yīng)溫度對化合物的合成也有一定影響。專利報道的溫度為100 ℃。因此,進一步考察不同反應(yīng)溫度對中間體6收率的影響,結(jié)果顯示,溫度超過100 ℃時,反應(yīng)體系雜質(zhì)多,原料大部分未反應(yīng)完全;溫度為80~100 ℃時,反應(yīng)體系雜質(zhì)減少,原料大部分反應(yīng)完全;溫度為70~80 ℃時,反應(yīng)體系干凈,原料幾乎反應(yīng)完全,產(chǎn)物純化簡單,收率高,因此,選擇反應(yīng)溫度為70~80 ℃,具體結(jié)果見表3。

表3 反應(yīng)溫度對中間體6收率的影響
綜上所述,在該反應(yīng)中,選擇反應(yīng)溶劑為1,4-二氧六環(huán)/水,碳酸鉀為堿,反應(yīng)溫度為70~80 ℃,中間體6收率可達到81%。
2.3 中間體7的合成分析
本研究對已有的合成路線進行了優(yōu)化,在合成中間體7時,將羧酸制成酰氯,再與氘代甲胺進行酰胺化反應(yīng)。本研究首先嘗試了原研專利的合成方法,并對其進行了優(yōu)化,嘗試了2種氯代試劑及堿對酰化反應(yīng)的影響。實驗結(jié)果顯示,采用原研專利的合成方法,以草酰氯為氯代試劑,N,N-二異丙基乙胺為堿時,反應(yīng)時間較長,反應(yīng)體系較雜,需要柱層析進行純化,收率低;使用氯化亞砜(SOCl2)為氯代試劑,三乙胺(TEA)為堿時,反應(yīng)快,雜質(zhì)少,只需要通過打漿對產(chǎn)物進行純化。因此,本文確定以SOCl2為氯代試劑,TEA為堿,收率為81%(表4, Entry 2)。
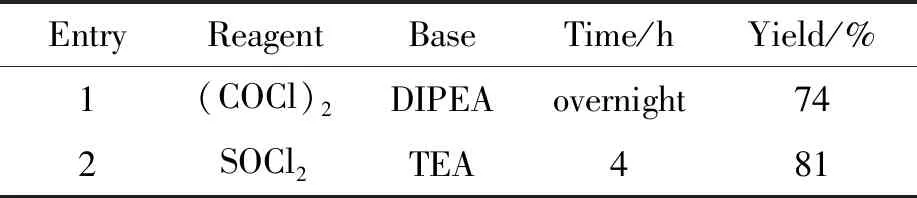
表4 氯代試劑及堿對中間體收率的影響
本文以1-溴-2-甲氧基-3-硝基苯為起始原料,通過還原、偶聯(lián)、芳基化和酰胺化4步反應(yīng),確立了有效合成BMS986202的反應(yīng)路線,并對反應(yīng)路線中的關(guān)鍵工藝條件進行了優(yōu)化。通過1H NMR, MS(ESI)對目標(biāo)產(chǎn)物BMS986202進行了表征,總收率達85%。合成BMS986202的工藝路線穩(wěn)定,反應(yīng)過程溫和,操作簡便且收率較高,可為進一步進行工業(yè)化生產(chǎn)打下良好基礎(chǔ)。
猜你喜歡
商品與質(zhì)量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀(jì)智能(數(shù)學(xué)備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛(wèi)生(2015年12期)2015-11-10 05:13:40
現(xiàn)代企業(yè)(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
