基于指紋圖譜結合化學模式識別的精天顆粒質量評價
2025-02-15 00:00:00趙偉陳樹和閆斌鄭瓊芳張偉欣巴元明
中國藥房 2025年3期



關鍵詞精天顆粒;指紋圖譜;超高效液相色譜法;層次聚類分析;主成分分析;正交偏最小二乘-判別分析;質量評價
慢性腎臟病(chronickidneydisease,CKD)是一種起病隱匿、治療周期長且患病率在數十年內顯著升高的疾病,現已成為全球發病率及病死率增長最快的疾病之一[1]。目前,化學藥治療CKD存在高血鉀、血肌酐升高等副作用,而中醫藥治療CKD的不良反應較少,且與化學藥聯合治療時,可減少化學藥的劑量和不良反應[2]。精天顆粒是湖北省中醫院腎病科巴元明教授在長期治療CKD的臨床實踐中凝練而成的經驗方,由酒黃精、紅景天、茯苓、豬苓、澤瀉、桂枝、熟大黃、穿山龍、白術、炙甘草10味藥組成。方中,酒黃精為君藥,可養陰潤肺、滋腎填精;紅景天為臣藥,可補氣清肺、益智養心;茯苓、豬苓、澤瀉、桂枝、白術可利水滲濕、溫陽化氣,熟大黃可瀉下、活血化瘀,穿山龍可祛風除濕、舒筋活血,共為佐藥;炙甘草為使藥,可顧護胃氣,使攻不傷胃,并調和諸藥;全方共奏益氣養陰、利水瀉濁之功效,用于氣陰兩虛、水濁內停所致的CKD3~4期患者。
本課題組擬將該方開發為醫療機構制劑。目前,在精天顆粒的質量標準草案(正在注冊申報)中,除了對其粒度、水分、溶化性等關鍵物理特性進行檢查外,還通過薄層色譜法成功鑒別了茯苓、熟大黃和大黃酸的特征性斑點。在含量測定項下,該草案選取了紅景天苷作為檢測成分,以評估臣藥的質量。除此之外,該草案尚未對處方中的其他藥材進行質控。因此,為了更好地控制精天顆粒的質量,有必要建立一種全面、高效、快速的檢測方法。指紋圖譜是一種全面且客觀的中藥質量評價工具,通過結合指紋圖譜與化學模式識別技術,如層次聚類分析(hierarchicalclusteranalysis,HCA)、主成分分析(principalcomponentanalysis,PCA)、正交偏最小二乘-判別分析(orthogonalpartialleastsquares-discriminantanalysis,OPLS-DA)等,可以更真實地反映中藥質量的差異,并揭示中藥復雜成分間的相互作用規律,這些方法已廣泛應用于中藥及其復方制劑的質量評價[3]。本研究擬采用超高效液相色譜(UPLC)法建立精天顆粒的指紋圖譜,并結合化學模式識別技術全面、系統地評價該制劑的質量,以期為該制劑的質量評價及控制提供科學依據。
1 材料
1.1 主要儀器
本研究所用主要儀器包括WayealLC3600系列UPLC儀(安徽皖儀科技股份有限公司)、AS220.R2型萬分之一天平(美國Ohaus公司)、ES225SM-DR型十萬分之一分析天平(瑞士Precisa公司)、KQ-500VDE型雙頻數控超聲波清洗器(昆山市超聲儀器有限公司)等。
1.2 主要藥品與試劑
酒黃精、紅景天、茯苓、豬苓、澤瀉、桂枝、熟大黃、穿山龍、白術、炙甘草飲片購于湖北辰美中藥有限公司、黃岡金貴中藥產業發展有限公司、河北楚風中藥飲片有限公司、保和堂(亳州)制藥有限公司、甘肅九州天潤中藥產業有限公司等,經湖北省中醫院藥事部陳樹和主任藥師鑒定均為真品。將不同廠家不同批次飲片進行隨機組合,再按照精天顆粒制備工藝制成顆粒,其中10批精天顆粒由湖北省中醫藥研究院自制,批號依次為20240801(S1)、20240802(S2)、20240803(S3)、20240804(S4)、20240805(S5)、20240806(S6)、20240807(S7)、20240808(S8)、20240809(S9)、20240810(S10);3批精天顆粒為勁牌持正堂藥業有限公司制備,批號分別為C0000230901(S11)、C0000230902(S12)、C0000230903(S13),以上規格均為18g/袋。
5-羥甲基糠醛對照品(批號111626-202316,純度98.4%)、紅景天苷對照品(批號110818-202210,純度99.7%)、綠原酸對照品(批號110753-202119,純度96.3%)、肉桂酸對照品(批號110786-202305,純度98.8%)、甘草酸銨對照品(批號110731-202122,純度94.4%)、大黃酸對照品(批號110757-201607,純度96.0%)、大黃素對照品(批號110756-201913,純度96.0%)、甘草次酸對照品(批號110723-202316,純度99.6%)、大黃酚對照品(批號110796-201922,純度99.4%)、蘆薈大黃素對照品(批號110795-201710,純度98.3%)均購自中國食品藥品檢定研究院;乙腈、磷酸均為色譜純,其余試劑均為分析純,水為超純水。
2 方法與結果
2.1 色譜條件
以Luna?OmegaPolarC18(150mm×2.1mm,1.6μm)為色譜柱,以乙腈(A)-0.2%磷酸溶液(B)為流動相進行梯度洗脫(0~3min,5%A→8%A;3~15min,8%A→15%A;15~50min,15%A→26%A;50~60min,26%A→42%A;60~68min,42%A→44%A;68~73min,44%A→70%A;73~76min,70%A→5%A;76~80min,5%A);進樣量為1μL;流速為0.2mL/min;柱溫為30℃;檢測波長為265nm。
2.2 溶液的制備
2.2.1 供試品溶液的制備
取精天顆粒適量,研細,精密稱定2g,置具塞錐形瓶中,精密加入甲醇20mL,稱質量,超聲(功率250W,頻率45kHz)處理30min;放冷后再次稱質量,用甲醇補足減失的質量;濾過,濾液以0.22μm微孔濾膜過濾,即得供試品溶液。
2.2.2 對照品溶液的制備
分別精密稱取5-羥甲基糠醛、紅景天苷、綠原酸、肉桂酸、大黃酸、甘草酸銨、大黃素、甘草次酸、大黃酚、蘆薈大黃素對照品適量,加甲醇制成一定濃度的單一對照品貯備液;分別取上述各單一對照品貯備液適量,置于同一容量瓶中,加甲醇定容,制成質量濃度分別為49.00、103.50、26.52、18.96、76.96、25.97、12.62、9.68、13.22、20.16μg/mL的混合對照品溶液。
2.2.3 單味飲片供試品溶液的制備
按處方比例分別稱取10味飲片,再按精天顆粒制備工藝制樣,得各單味飲片樣品;取各單味飲片樣品,按“2.2.1”項下方法處理,即得單味飲片的供試品溶液。
2.3 方法學考察
2.3.1 精密度試驗
取精天顆粒(S11)適量,按“2.2.1”項下方法制備供試品溶液,按“2.1”項下色譜條件連續進樣測定6次,記錄峰面積。以16號峰(峰形較好且響應值較高)為參考峰,計算得各共有峰相對保留時間和相對峰面積的RSD均不大于2.9%(n=6),表明該方法精密度良好。
2.3.2 穩定性試驗
取精天顆粒(S11)適量,按“2.2.1”項下方法制備供試品溶液,分別于室溫條件下放置0、2、4、8、12、24h時按“2.1”項下色譜條件進樣測定,記錄峰面積。以16號峰為參考峰,計算得各共有峰相對保留時間和相對峰面積的RSD均不大于3.8%(n=6),表明在室溫下24h內供試品溶液穩定。
2.3.3 重復性試驗
取精天顆粒(S11)適量,按“2.2.1”項下方法平行制備6份供試品溶液,按“2.1”項下色譜條件分別進樣測定,記錄峰面積。以16號峰為參考峰,計算得各共有峰相對保留時間和相對峰面積的RSD均不大于4.4%(n=6),表明該方法重復性良好。
2.4 精天顆粒指紋圖譜的建立
取13批精天顆粒,分別按“2.2.1”項下方法制備供試品溶液,按“2.1”項下色譜條件進樣測定,得到13批樣品的UPLC圖,再導入《中藥色譜指紋圖譜相似度評價系統(2012版)》軟件進行分析,設定樣品S13的圖譜為參照圖譜、時間窗寬度為0.1min,采用中位數法,結合多點校正并進行Mark峰匹配,生成精天顆粒對照指紋圖譜(R)及13批樣品的UPLC疊加指紋圖譜(圖1)。由對照指紋圖譜標定出25個共有峰;相似度分析結果顯示,13批樣品指紋圖譜與對照指紋圖譜的相似度為0.955~0.996,均在0.9以上,說明各批次間樣品具有高度相似性,各批次精天顆粒的制備工藝較為穩定,質量一致性較好。
2.5 精天顆粒共有峰的指認與歸屬
取按“2.2.3”項下方法制備的單味飲片供試品溶液,按“2.1”項下色譜條件進樣檢測,得各單味飲片的UPLC圖。將該色譜圖與混合對照品溶液的UPLC圖(R1)、精天顆粒對照指紋圖譜(R)同時進行比對(圖2),對樣品中各共有峰進行指認,共指認出其中10個,分別是3號峰5-羥甲基糠醛、5號峰紅景天苷、8號峰綠原酸、15號峰肉桂酸、19號峰蘆薈大黃素、20號峰甘草酸銨、21號峰大黃酸、23號峰大黃素、24號峰甘草次酸、25號峰大黃酚。對各單味飲片對共有峰的貢獻進行統計,結果可得,熟大黃、紅景天、穿山龍、炙甘草、白術、酒黃精、豬苓、桂枝、茯苓、澤瀉貢獻的共有峰分別有14、13、6、6、4、3、3、3、2、2個,詳見表1。
2.6 精天顆粒指紋圖譜的化學模式識別分析
2.6.1 HCA
采用SPSS26.0軟件對13批精天顆粒中25個共有峰的峰面積進行HCA,通過組間聯接和平方歐氏距離法進行處理,結果如圖3所示。由圖3可知,當平方歐氏距離為15時,13批樣品被劃分為3類:S1、S5、S7、S11~S13聚為一類,S4、S6聚為一類,S2、S3、S8~S10聚為一類。
2.6.2 PCA
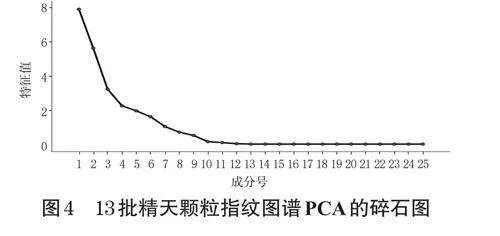
PCA屬于一種非監督的多元統計分析方法,能夠較為全面地反映多個指標成分原有的數據信息[4]。本研究以13批精天顆粒指紋圖譜中25個共有峰的峰面積為變量,將其導入SPSS26.0軟件進行PCA。以特征值>1作為篩選標準[5],得到7個主成分因子(圖4),其累計方差貢獻率達92.666%,表明該7個主成分因子可反映精天顆粒特征圖譜的主要信息。第1~7個主成分的因子載荷系數絕對值最高者依次為3號峰(5-羥甲基糠醛,0.914)、13號峰(0.934)、14號峰(0.974)、22號峰(0.903)、11號峰(-0.930)、12號峰(0.811)、15號峰(肉桂酸,0.965),這些成分的主要來源飲片為酒黃精、紅景天、熟大黃和桂枝。
2.6.3 OPLS-DA
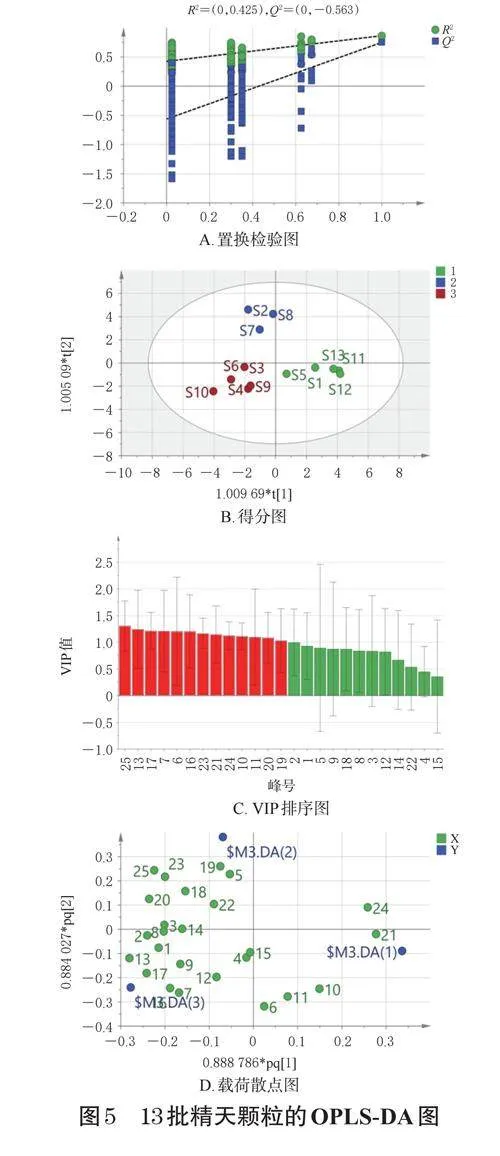
采用SIMCA14.1軟件對13批精天顆粒中25個共有峰進行OPLS-DA,以探究樣品間的差異性。結果顯示,模型的累計解釋率RX2(cum)、RY2(cum)和累計預測率Q2(cum)分別為0.538、0.888和0.764,均大于0.5,表明該模型具有良好的解釋和預測能力[6]。為驗證模型的穩健性,本研究進行了200次隨機置換檢驗,結果如圖5A所示。由圖5A可知,R2回歸線的Y軸截距為0.425,Q2回歸線的Y軸截距為-0.563,均低于原始值,證實了模型沒有過擬合,適合用于區分13批樣品間的差異。根據OPLS-DA得分,13批樣品可聚為3類(圖5B),其中S2、S7、S8聚為一類,S1、S5、S11~S13聚為一類,S3、S4、S6、S9、S10聚為一類,這一聚類結果與HCA結果存在部分差異。OPLS-DA常采用變量重要性投影(variableimportanceinprojection,VIP)評估每個共有峰的貢獻度,以VIP>1作為判定標準[7],從而識別組間差異的潛在關鍵因素。由圖5C可知,VIP>1的共有峰有13個,按VIP值從高到低依次為25號峰(大黃酚)、13號峰、17號峰、7號峰、6號峰、16號峰、23號峰(大黃素)、21號峰(大黃酸)、24號峰(甘草次酸)、10號峰、11號峰、20號峰(甘草酸銨)、19號峰(蘆薈大黃素)。這些色譜峰所對應的成分被認為是影響精天顆粒批次間差異的主要化學成分,即潛在的差異標志物。進一步分析顯示,這些差異標志物中,1個來自豬苓,1個來自穿山龍,3個來自炙甘草,5個來自紅景天,7個來自熟大黃(因6、7號色譜峰的來源飲片有多個,故此處合計值大于13),說明嚴格控制熟大黃、紅景天、炙甘草、穿山龍、豬苓飲片的質量是確保精天顆粒批次間質量穩定的關鍵。在載荷散點圖中,距離原點越遠的成分對樣品分類的影響越大。由圖5D可知,上述13個色譜峰均遠離原點,與圖5C結果一致,進一步證實了上述13個成分可能是導致樣品批次間差異的主要標志性成分。
3 討論
在供試品溶液的制備過程中,本研究考察了不同提取溶劑(甲醇、30%甲醇、50%甲醇、70%甲醇、乙醇、30%乙醇、50%乙醇、70%乙醇)、不同溶劑體積(10、20、50、100mL)、不同超聲時間(15、30、45min)對提取結果的影響,最終選擇了以20mL甲醇作為提取溶劑,超聲提取30min的供試品溶液制備方法。在色譜條件的優化方面,本研究對流動相(甲醇-0.2%磷酸溶液、乙腈-0.2%磷酸溶液、甲醇-水、乙腈-水)、柱溫(30、35、40℃)、進樣量(1、3、5μL)進行了考察,最終確定流動相為乙腈-0.2%磷酸溶液,柱溫為30℃,進樣量為1μL;用二極管陣列檢測器進行全波長(190~400nm)掃描,發現在265nm波長下,供試品溶液的響應信號較強且基線穩定,并能檢測出包含190~400nm波長范圍內的絕大部分色譜峰,故確定檢測波長為265nm。
本研究建立了13批精天顆粒的UPLC指紋圖譜,利用混合對照品溶液確認了10個特征峰的歸屬,指認出25個共有峰,并對這些共有峰進行了飲片歸屬。研究結果表明,13個不同批次樣品在化學成分上的一致性較好。在對13批精天顆粒進行指紋圖譜分析時,發現其相似度較高(0.955~0.996),表明樣品所采用的制備工藝和樣品質量較為穩定。HCA結果顯示,當平方歐氏距離為15時,樣品S1~S10被劃分為3個不同的類別,這可能與不同批次飲片的產地和質量差異有關;樣品S11~S13是采用相同批次的飲片在相同生產條件下制備的,當平方歐氏距離為5時,它們被單獨聚為一類,說明在保證飲片質量、生產條件一致的前提下,制劑質量較為一致。PCA和OPLS-DA的進一步分析結果揭示了多種成分的含量差異,這些成分主要來源于酒黃精、紅景天、熟大黃、炙甘草、桂枝、穿山龍和豬苓,故需要嚴格控制上述飲片的質量。HCA是一種非監督學習方法,其基于不同樣本之間的相似性進行分組,不依賴于預先定義的類別標簽;而OPLS-DA是一種有監督的判別分析方法,旨在通過最大化組間差異和最小化組內差異來識別不同組別之間的差異化合物。上述2種方法在算法設計和目的上存在本質區別,這可能是導致本研究中二者分組結果不一致的原因。
此外,鑒于所建立的指紋圖譜與茯苓、豬苓、澤瀉、穿山龍圖譜對應的共有峰數目較少,本研究還對文獻報道的茯苓中的活性成分茯苓酸[8],豬苓中的麥角甾醇[9],澤瀉中的23-乙酰澤瀉醇B、23-乙酰澤瀉醇C[10],穿山龍中的薯蕷皂苷[11]等成分進行了比對,但可能出于色譜柱不匹配、化學成分在樣品制備過程中發生轉化或者含量低于儀器的檢測限無法被檢出等原因,上述成分均不能在本研究色譜條件下被檢測到,后續可使用液相色譜-質譜聯用技術等方法對其進一步分析。
綜上所述,本研究所建立的指紋圖譜方法簡便、穩定、重復性良好,結合化學模式識別方法不僅適用于精天顆粒不同批次間的一致性評價,而且為全面控制精天顆粒的質量提供了重要參考依據。方中酒黃精、紅景天、熟大黃、穿山龍、豬苓、桂枝、炙甘草的質量是影響精天顆粒整體質量的關鍵。
