三維有序大孔徑介孔二氧化硅載體的仿生合成
2010-05-29 13:16:02汪志銀劉向農楊宇翔
浙江農林大學學報 2010年3期
關鍵詞:二氧化硅
曹 磊,汪志銀,劉向農,楊宇翔
(1.華東理工大學 化學與分子工程學院,上海 200237;2.浙江林學院 理學院,浙江 臨安 311300;3.揚州大學測試中心,江蘇 揚州 225002)
氯酚被廣泛用于木材防腐,也用于防銹劑、殺菌劑、殺蟲劑和除草劑等的生產中。如以DDT為原料中間體生產三氯殺螨醇(DTMC),以供果樹、棉花等經濟作物殺害蟲之用,所以,它們在環境中的存在還相當普遍。氯酚類農藥對生物組織具有較強的變性作用,強烈刺激皮膚、黏膜,并具有腐蝕性。氯酚的毒性隨其氯化程度的增加而增大,且難降解,能夠在環境中長期存在和積累[1]。實驗發現,用漆酶催化降解氯代農藥,降解效率高。但是由于漆酶在環境中容易失活,難以重復利用,制約了它的廣泛應用,所以酶的固定化技術是實現漆酶重復利用及提高其穩定性的有效手段[2-4]。介孔二氧化硅作為一種無機材料,具有較大的比表面積、較窄的孔徑分布和可以調變的孔徑,以及良好的化學穩定性、成本低廉、化學改性容易,且孔道內富含弱酸性羥基,因此,作為酶固定化的載體具有良好的發展前景[5-6]。根據李艷敬等[7]的研究可知,具有三維孔道結構的介孔二氧化硅載體要比具有一維孔道結構的二氧化硅載體更有利于酶的固定,這是因為三維孔道之間可以相互連接,有利于酶分子和底物分子的自由擴散。Pandya等[8]的研究發現介孔材料的孔徑太大或者太小都會造成催化效率的降低。當孔道直徑為酶分子直徑的2~5倍時,可以起到很好的催化降解效果。一般實驗室用于降解氯酚農藥的漆酶直徑約為2 nm,因此,合成孔徑為5~10 nm的具有三維孔道結構的介孔二氧化硅載體對于研究固定漆酶降解氯酚農藥具有十分重要的意義。本研究通過研究兩性生物表面活性劑椰油基甘氨酸鈉(YCS)與不同鏈長的陽離子表面活性劑烷基三甲基溴化銨(CnTAB,n=12,14,16,18)的混合體系,成功的制得了具有較大孔徑與比表面積的三維孔道的介孔二氧化硅載體,為進一步固定漆酶降解氯酚農藥的研究打下基礎。
1 實驗
1.1 實驗原料及儀器
正硅酸乙酯(TEOS),十二烷基三甲基溴化銨(C12TAB),十四烷基三甲基溴化銨(C14TAB),十六烷基三甲基溴化銨(C16TAB),十八烷基三甲基溴化銨(C18TAB),氨水,硫酸(H2SO4)(以上試劑皆為分析純,國藥集團化學試劑有限公司),椰油基甘氨酸鈉(YCS)(上海中獅科技有限公司)。
用日本理學2550D/maxVB/PC轉靶X射線衍射儀(Cu靶Kα線,λ=0.15406 nm)進行小角度衍射測量,以確定介孔二氧化硅的孔結構;用美國Micromeritics公司ASAP2405N物理吸附儀測定材料的吸附性能及孔結構,分別采用BET(Brunaure-Emmet-Teller)方程和BJH(Berret-Joyner-Halenda)模型計算樣品的比表面積、孔徑分布和孔體積;用日本JEOL JEM-3010型透射電鏡來觀察介孔二氧化硅的孔形狀及孔有序度;日本JEOL JSM-6360LV型掃描電鏡觀察介孔二氧化硅的表面形貌。
1.2 制備過程
將不同質量的C16TAB與YCS混合于30℃下溶于67.00 mL去離子水中,磁力攪拌到溶液變為澄清后加入5.00 mL硫酸,攪拌1 h后,滴加3.35 mL正硅酸乙酯(TEOS)。溶液繼續攪拌24 h,加入一定量的氨水,攪拌1 min。升溫至80℃老化24 h。將所得白色固體過濾,沖洗,室溫下干燥。組分的摩爾比為:xCnTAB ∶(1 - x)YCS ∶3.0 TEOS ∶18.7 H2SO4∶741 H2O ∶47 NH3H2O,其中 R=x/(1 - x)(x=0.9,0.8,0.7,0.6,0.5)。將所得白色粉末以2℃·min-1的速度升溫至550℃煅燒6 h。分別將1.232 g C12TAB,1.344 g C14TAB,1.568 g C18TAB與0.266 g YCS混合重復上述步驟,其摩爾組分為:0.8CnTAB ∶0.2 YCS ∶3.0 TEOS ∶18.7 H2SO4∶741 H2O ∶47 NH3H2O (n=12,14,16,18)。將 C12TAB與YCS按摩爾比為8∶2混合后制得的煅燒產物命名為C12YS,C14TAB與YCS按摩爾比為8∶2混合后制得的煅燒產物命名為C14YS,C16TAB與YCS按摩爾比為8∶2混合后制得的煅燒產物命名為C16YS,C18TAB與YCS按摩爾比為8∶2混合后制得的煅燒產物命名為C18YS。另外,稱取1.458 g C16TAB作為單一模板劑重復上述實驗,研究單一模板劑對產物結構的影響。該反應的摩爾組分為:0.8 C16TAB ∶3.0 TEOS ∶18.7 H2SO4∶741 H2O ∶47 NH3H2O。
2 結果與討論
2.1 熱重(TGA,thermo gravimetric analysis)-差熱(DTA,differential thermal analysis)分析
圖1為C16TAB+YCS按摩爾比為8∶2混合后制得樣品的TGA/DTA圖,由圖1可見,TGA曲線有3個失重區間。在252.3℃之前的第一失重段,DTA曲線有一弱吸收峰,對應吸附水的脫附,失重為3%;在252.3~500.0℃之間的第二失重段,DTA曲線有一強放熱峰,歸因于表面活性劑的完全分解和硅酸根多聚體中結合水的去除,此段的失重為31%;而在第三失重段500~700℃,DTA曲線有一弱放熱峰,此段的失重僅為3%,對應于無機骨架的進一步縮聚;當處于DTA曲線頂峰573.4℃溫度時,樣品孔道內表面結構羥基進一步締合,引起孔道的塌陷,以及原子重新聚結[9],因而造成介孔結構的消失。從圖1還可以看出,在550℃以上,質量失重只有1%,表明樣品經過550℃煅燒后,全部轉化為純的二氧化硅粉末。綜合上述因素,本研究中樣品的煅燒溫度選為550℃。
2.2 X射線衍射儀(XRD,X-ray diffractomer)分析
圖2為使用單一模板劑C16TAB與混合模板劑制得介孔二氧化硅煅燒前后的XRD圖。
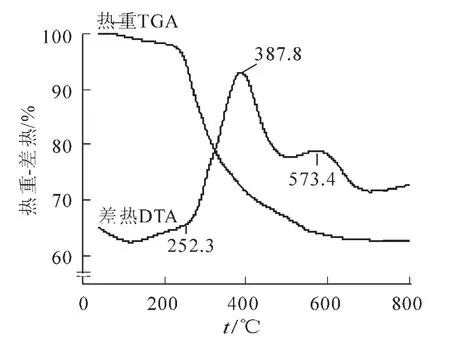
圖1 混合體系(C16TAB+YCS)的差熱熱重(R=8∶2)Figure1 Thermogravimetric and differential thermal analysis of mixed system (C16TAB+YCS)
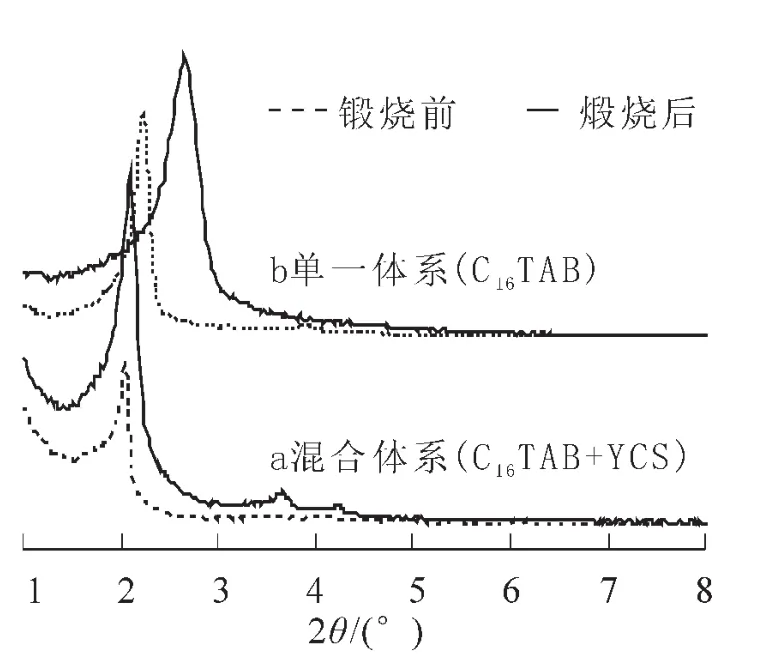
圖2 不同體系制得樣品的XRD圖Figure2 XRD patterns of mesoporous silica formed with different synthesis systems
從圖2a中我們可以看到,對于混合體系而言,所得樣品在煅燒前后都保持良好的有序度,煅燒后可以清楚的看到,除了主峰之外還存在2個明顯的弱衍射峰。且煅燒后樣品的有序度明顯增強,計算得d100僅為0.15。從圖2b中我們可以看到,對于單一體系而言,合成產物煅燒后的XRD峰發生了明顯的寬化,且向右發生了橫移,說明產物經過煅燒后有序度下降,同時,存在一定程度的孔徑收縮的現象;計算得d100為0.66,是使用混合表面活性劑得到產物的4倍。因此,使用混合體系制得產物煅燒后的有序度明顯高于單一體系,椰油基甘氨酸鈉(YCS)的加入提高了產物的有序度。另外,比較圖2還可以發現,混合體系制得產物經煅燒后的XRD峰更趨向于y軸,說明通過混合體系制得產物煅燒后的孔徑要大于單一體系,YCS在體系中起到了擴孔劑的作用。因此,使用混合體系有利于得到有序度更高且孔徑更大的介孔二氧化硅。
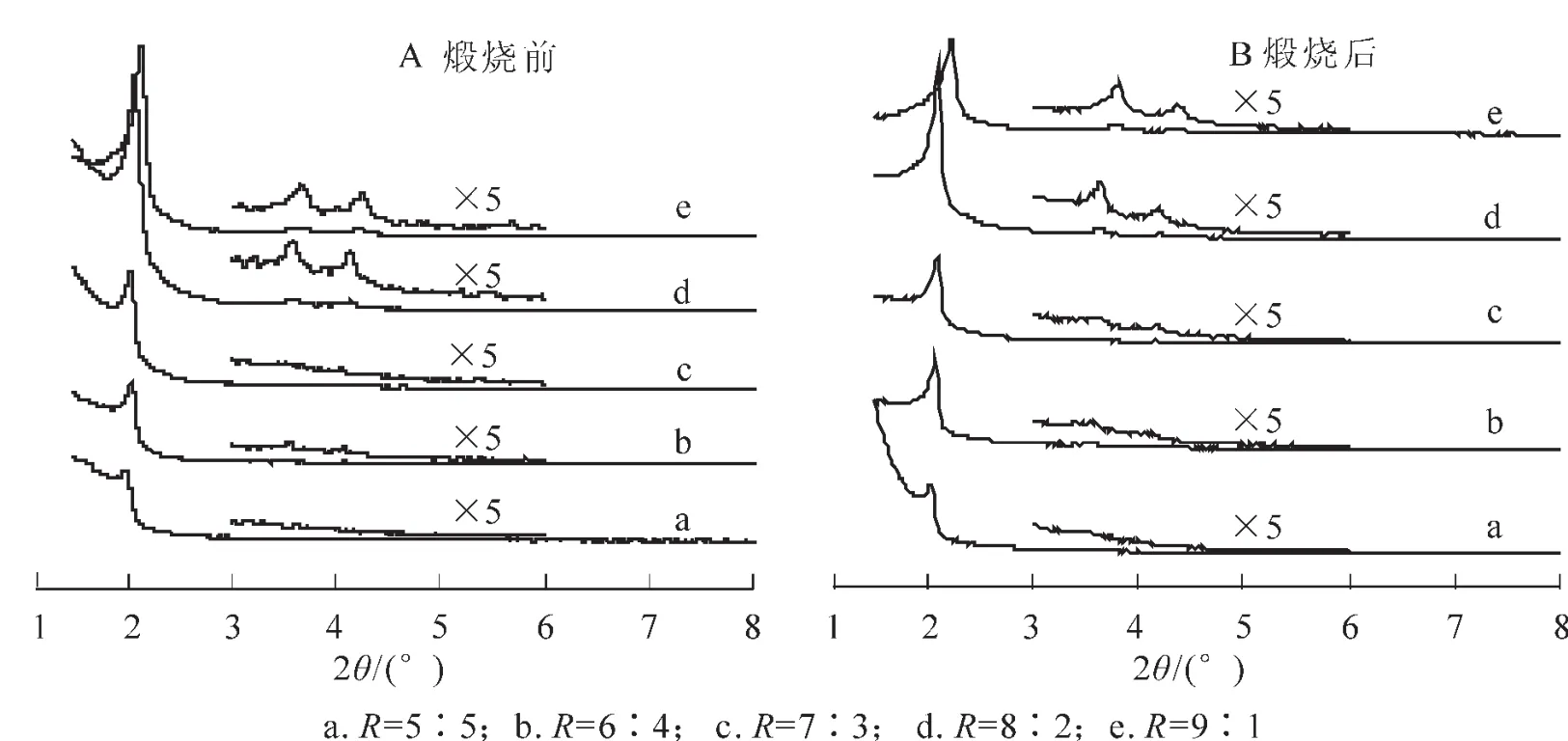
圖3 不同R值條件下合成樣品的XRD圖Figure3 XRD patterns of mesoporous silica formed with different R
圖3為按照不同摩爾組分混合2種表面活性劑制得樣品煅燒前后的XRD圖。如圖3所示,在不同的R值條件下,我們均可得到有序的介孔結構。其中,當R=8∶2時,Δd100=0.04為最小值(表1),產物的孔壁最為致密;而且其煅燒前后的XRD主峰最高,2個弱峰也清晰可見,這說明合成產物有序非常好。因此,當C16TAB與YCS按照摩爾比為8∶2混合時,得到合成產物的有序度最高。
圖4為不同鏈長陽離子表面活性劑(CnTAB,n=12,14,16,18)與YCS按照摩爾比為8∶2的條件下混合后所制得的樣品煅燒前后的XRD衍射圖。由圖4可以看出,樣品C12YS在煅燒前無明顯的衍射峰,但是煅燒后在2θ=2.18°處有1個強的衍射峰。樣品C14YS,C16YS,C18YS均在低角度區域存在一個強的晶面衍射峰,此外,在3°~5°區域還存在2個弱的吸收峰,且3個衍射峰的晶面間距的比值為1∶∶2,這與文獻報道的具有六方對稱特征的典型介孔分子篩MCM-41的特征衍射峰相符合。這表明我們所合成樣品中的無機骨架是具有MCM-41特征的六方介孔結構。樣品煅燒后的XRD峰高遠高于煅燒前,說明煅燒后樣品的有序度明顯增加。由表2可知,隨著碳鏈的增加,樣品煅燒前后晶胞的d100逐漸減小,孔壁變的更加致密;XRD曲線向左移動,產物的孔徑逐漸增加:與此同時,比表面積與孔體積也逐漸增加。根據親水-疏水平衡(HLB)原則,產生某一類的組裝時,表面活性劑的疏水部分(碳氫鏈長)和親水部分(頭基加上反離子)應該是平衡的[10]。同一類型的表面活性劑具有相同的頭基基團,疏水鏈的增加意味著需要更多的反離子與其締合才能保持平衡。由此催化了大量的硅物種在膠束表面縮聚,孔壁變得更加致密,因而產物在焙燒前后晶胞收縮最小。
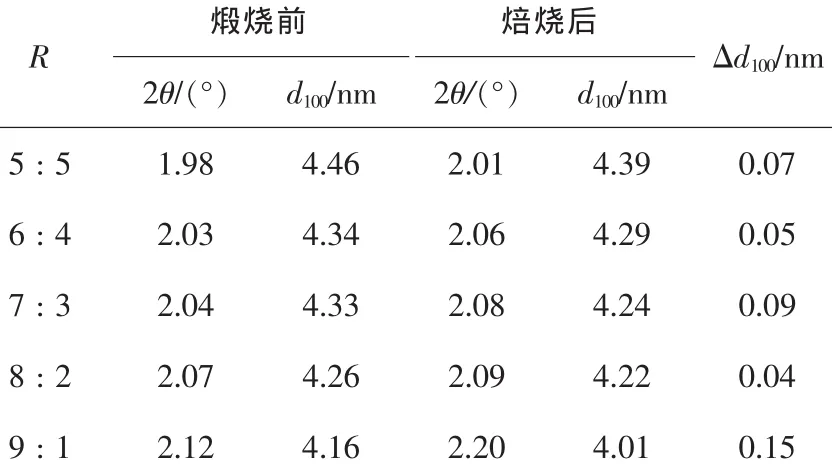
表1 不同R值條件下X-射線衍射數據指標化計算結果Table1 Index results of XRD data with different R
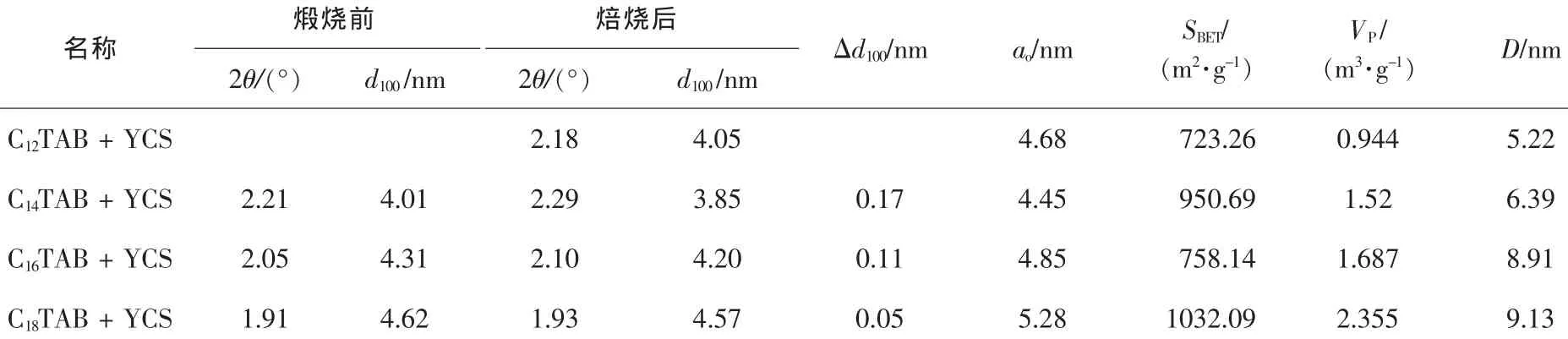
表2 不同的鏈長陽離子表面活性劑合成產物的孔結構參數Table2 Porous properties of samples with different chain length of cationic surfactants
從圖4還可以發現,當將C14TAB與YCS混合后制得煅燒產物的有序度最好。這是因為當不同鏈長的陽離子表面活性劑與YCS混合時,由于C14TAB與YCS具有相同的烷基鏈長,它們在空氣/水的界面上的堆積更加緊密。但是,當2種混合表面活性劑具有不同的鏈長時,相鄰分子之間具有較長鏈長的表面活性劑分子超出較短鏈長分子的那部分烷基鏈表現出熱運動的趨勢。這一熱運動干擾沿著鏈長向分子的極性頭基傳播,引起了較長鏈長分子所占區域的增加,導致混合分子的排列更加松散,產物的有序度降低[11]。
2.3 氮氣(N2)吸附-脫附測試分析
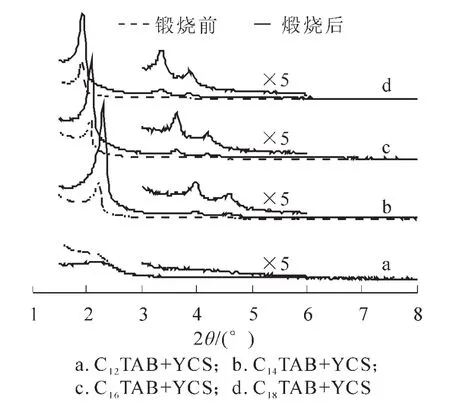
圖4 不同鏈長陽離子表面活性制得產物的XRD圖Figure4 XRD patterns of mesoporous silica formed with different chain-length of cationic surfactants
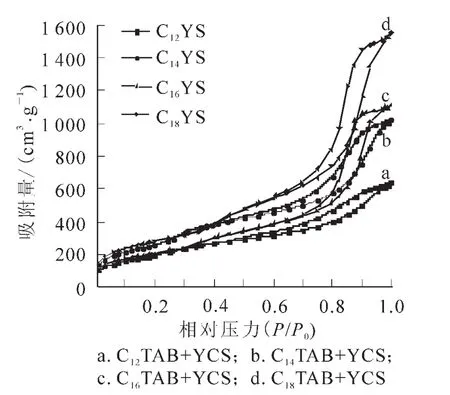
圖5 不同鏈長陽離子表面活性劑制得樣品的氮氣吸附等溫線Figure5 Nitrogen sorption/desorption isotherms of the calcined mesoporous silica synthesized by different chain length of cationic surfactants
圖5為所合成的不同介孔二氧化硅樣品的氮氣吸附-脫附等溫線和孔徑分布曲線圖。所有樣品的等溫線均呈現IUPAC(國際理論與應用化學聯合會)分類中的Ⅳ型吸附等溫線的特征,是典型的介孔材料吸附曲線,并且具有3個明顯區分的區域,即單層—多層吸附區域,毛細管濃縮區域及外表面多層吸附區域。在中壓段和接近飽和蒸氣壓時,均有一個H1類滯后環,且吸附分支和脫附分支幾乎平行,這說明合成產物具有大小均勻且形狀規則的柱狀孔道結構。中間段相對壓力的位置決定樣品孔徑的大小,吸附線變化的陡峭程度也是衡量介孔分布均一性的重要依據。一般認為:發生毛細管凝聚相對壓力靠后和具有大的滯后環,則說明樣品具有較大的孔徑。樣品C12YS,C14YS,C16YS和C18YS的吸附突躍位置依次向后推移,分別發生在P/P0=0.445,P/P0=0.448,P/P0=0.548和P/P0=0.55,且隨著鏈長的增加,吸附線變化的陡峭程度越大,這說明隨著鏈長的增加,合成產物的孔徑增加,且介孔孔徑分布趨于均勻化。
圖6為不同鏈長陽離子表面活性劑制得樣品的孔徑分布圖,樣品C12YS的孔徑主要分布在1.98~3.00 nm,3.00~4.33 nm和4.33~40.36 nm 3個范圍內,分別占到總粒子數的33.93%,16.25%和49.81%,平均孔徑為5.22 nm;樣品C14YS的孔徑主要分布在1.89~2.98 nm,3.33~4.19 nm和4.19~22.53 nm 3個范圍內,分別占到總粒子數的21.55%,14.57%和61.63%,平均孔徑為6.39 nm;樣品C16YS的孔徑主要分布在1.83~5.22 nm和5.22~21.86 nm 2個范圍內,分別占到總粒子數的29.01%和70.02%,平均孔徑為8.91 nm;樣品C18YS的孔徑主要分布在1.84~5.24 nm和5.24~23.31 nm 2個范圍內,分別占到總粒子數的30.12%和68.22%,平均孔徑為9.13 nm。根據以上分析可知,隨著陽離子表面活性劑鏈長的增加,產物的平均孔徑逐漸增大。與XRD的分析結果一致。合成產物具有非單一的孔徑分布范圍可能跟產物生成相互連通的三維六方孔道有關[12]。樣品C12YS,C14YS具有3個孔徑分布范圍,表明除了單孔徑及三連通孔徑之外,還存在少量雙連通孔徑。樣品C16YS,C18YS均具有2個孔徑分布范圍,說明產物主要為單孔徑及三連通孔徑。根據XRD結果計算發現產物的ao值小于其對應的平均孔徑,這一結果也說明產物中可能存在孔道連通的情況。根據文獻可知[13],當使用C16TAB作為結構導向劑時,合成產物的平均孔徑在2.50 nm作用,明顯小于混合體系制得產物的平均孔徑,因此,使用混合體系更有利于得到大孔徑三維孔道排列產物。
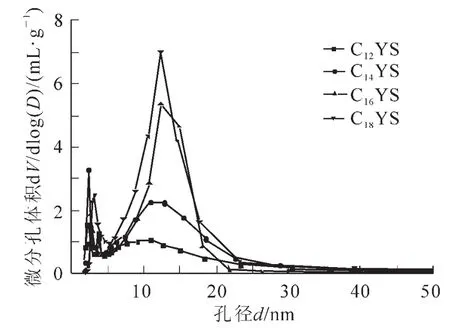
圖6 不同鏈長陽離子表面活性劑制得樣品的孔徑分布圖Figure6 Pore size distribution of the calcined mesoporous silica synthesized by different chain length of cationic
2.4 電子掃描電鏡(SEM,scanning electron microscopy)分析
圖7為不同鏈長陽離子表面活性劑與YCS混合后制得樣品煅燒后的SEM圖。樣品C12YS的形貌呈不規則的塊狀,由圖7b可以看到,在塊狀顆粒的表面出現了少量大小不一的球形顆粒的團聚。樣品C14YS的形貌呈塊狀和蠶豆狀的混合形貌,由圖7 d可以看到,在塊狀顆粒的表面出現大量大小形貌均一的球形顆粒的團聚。樣品C16YS的形貌以蠶豆狀為主,并伴有少量球形顆粒。樣品C18YS的形貌呈長短粗細不一的棒狀,仔細觀察可以發現是這些短棒是由無數的層狀結構堆積而成的。

圖7 不同鏈長陽離子表面活性劑制得樣品的SEM圖Figure7 SEM of calcined samples prepared using mixed surfactants with different cationic carbon chain lengths
離子表面活性劑的堆積參數被廣泛的用于預測和解釋最終的介孔結構[14]。g值的計算很簡單,但是卻有著重要的意義和指導作用:g=V/(aol)。這里,V是表面活性劑疏水鏈加上位于鏈間的所有的助溶劑(有機分子)的總體積,a0是位于水-膠束表面的有效親水頭基基團的面積,l是疏水基碳氫鏈長度。將g值和預期的介孔相序列做函數,當g<1/3時,體系形成球形膠團;當1/3<g<1/2,體系形成棒狀膠團;當1/2<g<1,體系將形成具有層狀膠團。根據文獻可知[15],對于試驗中使用到的陽離子表面活性劑C12TAB,C14TAB,C16TAB,C18TAB而言,隨著其疏水鏈長的增加,g值增加。另一方面,在混合體系中,由于兩性表面活性劑的氨基端帶有部分的正電荷,導致混合體系在膠束的自組織過程中,由于帶有正電荷的氨基端與相鄰排列的陽離子表面活性劑的疏水鏈部分產生靜電排斥,二者之間的排列間隙增加,陽離子表面活性劑的疏水鏈部分發生一定程度的卷曲,隨著陽離子表面活性劑疏水鏈長度的增加,由C12依次增加到C18的過程中,碳原子數的增加意味著其對應的疏水鏈的卷曲程度逐漸增強,總體積V值逐漸變大,使得g值也變大。另外,根據親水親油平衡(HLB),陽離子表面活性劑疏水鏈越長,其反離子的締合常數越大,利于形成更長的棒狀膠束[10]。因此,在三者的共同作用下,膠團的形貌由塊狀形貌向橢球形形貌轉變,最后變成棒狀形貌。
2.5 透射電鏡(TEM,transmission electron microscopy)分析
圖8是不同鏈長陽離子表面活性劑與YCS混合后制得樣品煅燒后的TEM圖。從圖8可以看出,無論使用何種鏈長的陽離子表面活性劑,制得的產物均為三維六方相的結構。三維六方相結構屬于立方相和六方相的混合相。使用不同的混合體系可以得到具有不同共生結構的三維六方產物。樣品C12YS具有不同孔道方向的立方相和六方相的混合相(圖8a);樣品C14YS具有P63/mmc-Ia3b的共生結構(圖8b);樣品C16YS則具有P63/mmc-Im3m的共生結構(圖8c);而樣品C18YS具有P63/mmc-Fm3m的共生結構(圖8d)[16]。粗略測量樣品C16YS和C18YS的兩孔心間距分別為4.53和5.24 nm,與由小角度X射線散射(SAXS,small angle X-ray scattering)的數據計算所得的a0=4.85 nm和a0=5.28 nm基本一致。
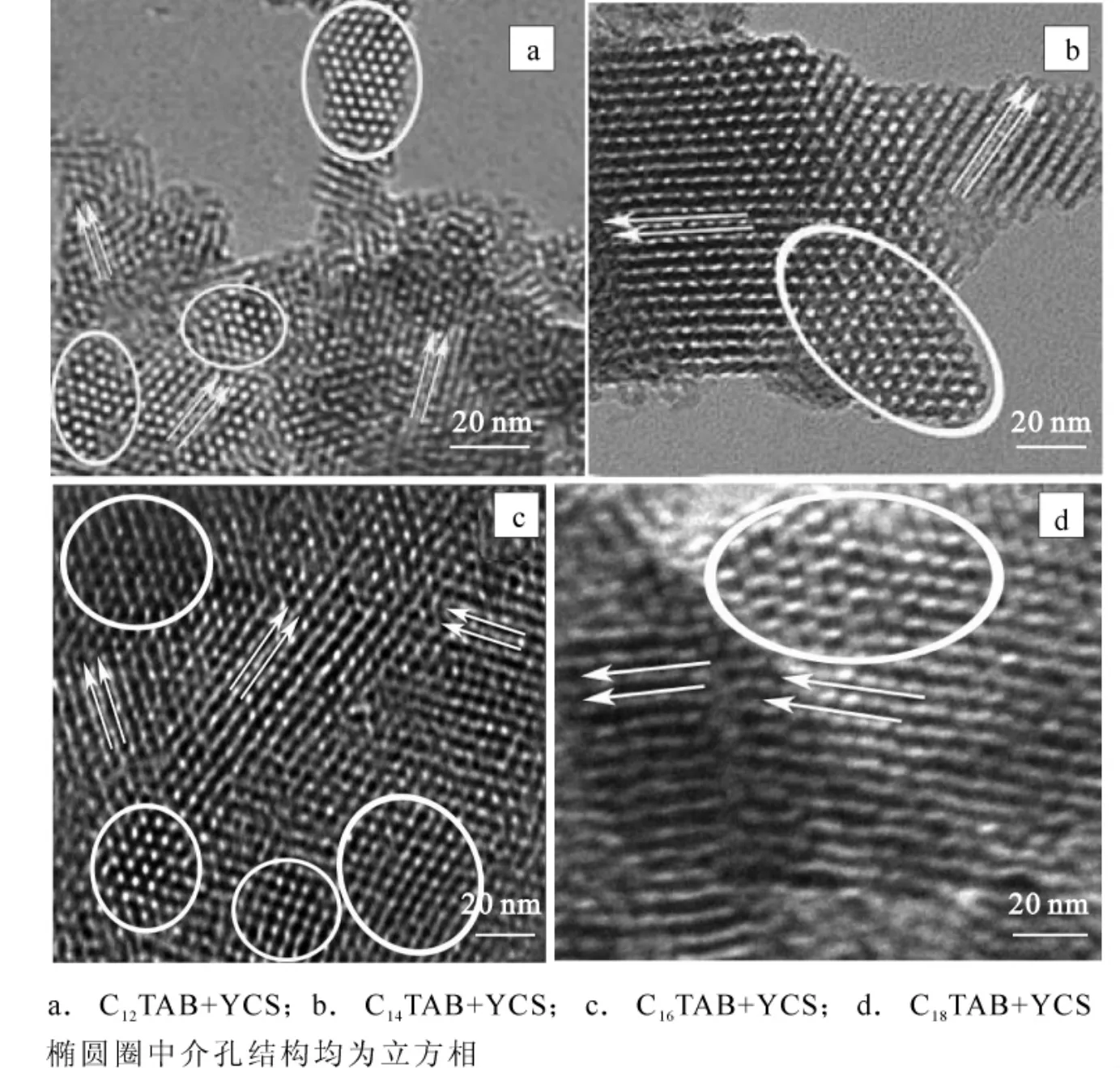
圖8 不同鏈長陽離子表面活性劑制得樣品的TEM圖Figure8 TEM of calcined samples prepared using mixed surfactants with different cationic carbon chain lengths
在酸性條件下,介孔二氧化硅的合成機制屬于S+X-I+路徑[17-20]。其中,S+和I+是陽離子表面活性劑和前驅體,X-是鹵素離子(Cl-,Br-和 I-),SO42-,NO3-等等。在強酸性介質中,S+X-I+相互作用最終是通過庫侖力實現的,或者更確切地說是雙層氫鍵的相互作用。然而,在混合膠束體系中,兩性表面活性劑YCS帶有負電荷的陰離子頭基基團[21]首先跟CTA+通過靜電相互作用相互結合,一方面導致了體系中正電荷數量的下降;另一方面,由于體系中存在一定數量的負電荷,靜電排斥作用導致反離子跟陽離子表面活性劑的結合力降低。而且由于硫酸本身的締合常數較小[22],導致膠束與硅物種結合較弱,在膠束表面縮聚的硅物種數量降低,孔壁變薄。在煅燒的過程中,相鄰孔道之間的孔壁發生塌陷,從而形成孔道連通的情況。
3 結論
跟單一體系十六烷基三甲基溴化銨相比,使用十六烷基三甲基溴化銨(C16TAB)與椰油基甘氨酸鈉(YCS)的雙組份體系制得有序度更高且孔徑更大的介孔二氧化硅。當混合體系中C16TAB與YCS的摩爾比為8∶2時,得到產物的有序度最高。不同鏈長的烷基三甲基溴化銨 (n=12,14,16,18)與YCS混合,成功制得具有不同形貌,孔徑分布(5~10 nm)以及比表面積(>700 m2·g-1)的三維介孔二氧化硅。隨著陽離子表面活性劑烷基鏈長的增加,煅燒產物的孔壁變的更加致密,比表面積、孔體積和孔徑增加。
[1]AGGELIS G,EHALIOTIS C,NERUD F,et al.Evaluation of white-rot fungi for detoxification and decolofizafion of effluents from the green olive debittering process [J].Appl Microbiol Biotechnol,2002,59:353 - 358.
[2]DURAN N,ROSA M A,D’ANNIBALE A,et al.Applications of laccases and tyrosinases(phenoloxidases) immobilized on different supports:a review [J].Enzyme Micro Technol,2002,31 (7):907 - 93l.
[3]WAN Yunyang,DU Yumin,SHI Xiaowen,et al.Immobilization an d characterization of laccase from Chinese Rhus vemicifera on modified chitosan [J].Process Biochem,2006,41 (6):l378 - l382.
[4]JOLIVALT C,BRENON S,CAMINADE E,et al.Immobilization of laccase from trametes versicolor on a modified PVDF microfiltration membrane:characterization of the grafted support and application in removing a phenylurea pesticide in wastewater[J].J Membr Sci,2000,180 (1):103 - 113.
[5]WU Jinchuan,LEE S S,MAHMOOD M M B,et al.Enhanced activity and stability of immobilized lipases by treatment with polar solvents prior to lyophilization [J].J Mol Catal B Enzymatic,2007,45:108 - 112.
[6]張樹江,高恩麗,夏黎明.固定化漆酶對二氯酚的脫氯作用[J].化工學報,2006,57(2):359-362.ZHANG Shujiang,GAO En’li,XIA Liming.Dechlorination of dichlorophenol in waste water by immobilized laccase [J].J Chem Ind Eng China,2006,57 (2):359 - 362.
[7]李艷敬,周國偉,邢凡勤.介孔材料中酶固定化的影響[J].應用化工,2008,37(6):688-691.LI Yanjing,ZHOU Guowei,XING Fanqin.Influence of immobilized enzyme in the mesoporous material[J].Appl Chem Ind,2008,37 (6):688- 691.
[8]YADAV G D,JADHAV S R.Synthesis of reusable lipases by immobilization on hexagonal mesoporous silica and encapsulation in calcium alginate:transesterifieation in non-aqueous medium [J].Microporous Mesoporous Mater,2005,86:215-222.
[9]ILER R K.The Chemistry of Silica[M].New York:Wiley&Sons,1979:389 - 412.
[10]LIN Hongping,MOU Chungyuan.Structural and morphological control of cationic surfactant-templated mesoporous silica[J].Accounts Chem Res,2002,35:927 - 935.
[11]PATIST A,CHHABRA V,PAGIDIPATI R,et al.Effect of chain length compatibility on micellar stability in sodium dodecyl sulfate/alkyltrimethylammonium bromide solutions [J].Langmuir,1997,13:432 - 434.
[12]FAN Jie,YU Chengzhong,WANG Liming,et al.Mesotunnels on the silica wall of ordered SBA-15 to generate three-dimensional large-pore mesoporous networks [J].J Am Chem Soc,2001,123:12113 - 12114.
[13]LIN Hongping,KAO Chiapei,MOU Chungyuan,et al.Counterion effect in acid synthesis of mesoporous silica materials[J].J Phys Chem B,2000,104:7885 - 7894.
[14]HUO Qisheng,MARGOLESE D I,STUCKY G D.Organization of organic molecules with inorganic molecular species into nanocomposite biphase arrays [J].Chem Mater,1996,8:1147 - 1160.
[15]YANG Yuxiang,YING Haiping,SHAO Jianguo,et al.A study on the effect of template chain length on the synthesis of mesoporous silica in an acidic condition [J].J Am Ceram Soc,2007,90 (11):3460 - 3467.
[16]SHERIF A E S,TAKAAKI H,FUJIO M.Design of highly stable,ordered cage mesostructured monoliths with controllable pore geometries and sizes [J].Chem Mater,2005,17:3137 - 3145.
[17]HUO Qisheng,MARGOLESE D I,CIESLA U,et al.Generalized synthesis of periodic surfactant/inorganic cooperative materials [J].Nature,1994,368:317 - 321.
[18]KRESGE C T,LEONOWICZ M E,ROTH W J,et al.Oredered mesoporous molecular sieves synthesized by a liquidcrystal-template mechanism [J].Nature,1992,359:710 - 712.
[19]BECK J S,VARTULI J C,ROTH W J,et al.A new family of mesoporous molecular sieves prepared with liquid crystal templates [J].J Am Chem Soc,1992,114:10834 - 10843.
[20]YANAGISAWA T,SHIMIZU T,KURODA K,et al.The preparation of alkyltrimethylammonium-kanemite complexes and their conversion to microporous materials [J].Bull Chem Soc Jpn,1990,63:988 - 992.
[21]YANG Yuxiang,HUANG Zheng,DENG Wenjing,et al.Synthesis of mesoporous silica templated by sodium N-dodecyl glycine [J].Microporous Mesoporous Mater,2008,110:267 - 276.
[22]YANG Hong,VOVK G,SOKOLOV I,et al.Synthesis of mesoporous silica spheres under quiescent aqueous acidic conditions[J].J Mater Chem,1998,8:743 - 750.
猜你喜歡
保定學院學報(2022年6期)2022-12-01 09:54:24
能源工程(2021年6期)2022-01-06 02:04:30
陶瓷學報(2020年5期)2020-11-09 09:23:00
天津醫科大學學報(2019年3期)2019-08-13 06:53:02
中成藥(2018年2期)2018-05-09 07:19:43
上海建材(2017年5期)2018-01-22 02:58:50
天津農學院學報(2016年2期)2016-12-01 05:40:05
航天制造技術(2016年6期)2016-05-09 08:32:39
廣西林業科學(2016年2期)2016-03-20 05:53:37
江蘇科技大學學報(自然科學版)(2015年4期)2015-12-17 12:42:40
