大孔吸附樹(shù)脂提取分離去甲基萬(wàn)古霉素的工藝研究
2010-06-04 08:34:28王健,林毅,王海燕等
化學(xué)與生物工程 2010年5期
去甲基萬(wàn)古霉素是我國(guó)唯一一個(gè)自主研制并成功應(yīng)用于臨床的糖肽類(lèi)抗生素,其結(jié)構(gòu)比萬(wàn)古霉素在亮氨酸部位少 1 個(gè)甲基,見(jiàn)圖1。

圖1 去甲基萬(wàn)古霉素結(jié)構(gòu)式
去甲基萬(wàn)古霉素具有和萬(wàn)古霉素相同的抗菌譜和相似的抗菌活性,對(duì)耐青霉素G及多種抗生素的金葡球菌、表球菌、溶血性鏈球菌、草綠色鏈球菌、肺炎球菌及腸球菌等均有強(qiáng)大的抗菌作用,對(duì)厭氧的難辨梭狀芽孢桿菌亦有較好的抗菌作用,對(duì)炭疽桿菌、白喉?xiàng)U菌等亦敏感,屬速效殺菌藥[1]。
目前萬(wàn)古霉素的分離純化方法有大孔樹(shù)脂吸附分離法[2]、沉淀法[3]、色譜制備法[4]、磁性親和吸附法[5]、雙水相萃取法[6]、反膠團(tuán)萃取法[7]等,也可將其中幾種方法綜合運(yùn)用[8]。色譜制備法、磁性親和吸附法成本較高,雙水相萃取法、沉淀法、反膠團(tuán)萃取法影響因素較復(fù)雜,提取率不高。大孔樹(shù)脂吸附分離法較其它方法具有一定的優(yōu)越性:(1)影響因素少,工序簡(jiǎn)單,操作方便;(2)提取率高,去除雜質(zhì)能力強(qiáng);(3)適用于工業(yè)化生產(chǎn)。由于去甲基萬(wàn)古霉素和萬(wàn)古霉素物化性質(zhì)極為接近,因此,作者采用大孔樹(shù)脂吸附分離法提取發(fā)酵液中的去甲基萬(wàn)古霉素,并優(yōu)化了相關(guān)的工藝參數(shù)。
1 實(shí)驗(yàn)
1.1 試藥、試劑與儀器
發(fā)酵液,華北制藥新藥公司。
去甲基萬(wàn)古霉素對(duì)照品,SIGMA公司;乙醇、鹽酸、氫氧化鈉均為工業(yè)級(jí);HZ801型、HZ802型、HZ803型、HZ820型、HZ816型樹(shù)脂,上海華震公司;NKA型、S-8型樹(shù)脂,南開(kāi)大學(xué)化工廠;XAD-16型樹(shù)脂,上海羅門(mén)哈斯化工公司。
高效液相色譜儀,Waters公司。
1.2 預(yù)處理
1.2.1 發(fā)酵液預(yù)處理
取發(fā)酵液加2 mol·L-1鹽酸調(diào)pH值至3.5,然后加入一定量的助濾劑過(guò)濾,濾液作為上柱液備用。
1.2.2 樹(shù)脂的預(yù)處理/再生
新(再生)樹(shù)脂首先用4 BV工業(yè)酒精浸泡24 h,用純化水洗至無(wú)醇味;再用3 BV 2 mol·L-1NaOH溶液攪拌浸泡3 h,用純化水洗至中性;然后用3 BV 2 mol·L-1HCl溶液攪拌浸泡3 h,用純化水洗至中性;再用3 BV 2 mol·L-1NaOH溶液浸泡3 h,用純化水洗至中性,待用。
1.3 吸附條件研究
1.3.1 樹(shù)脂的篩選
取已處理好的8種樹(shù)脂,分別裝入50 mL樹(shù)脂柱(R∶H=1∶5)中,取同一批號(hào)的去甲基萬(wàn)古霉素上柱液,分別在常溫下以1.5 BV·h-1的流速上樣,保證樹(shù)脂吸附飽和或過(guò)飽和,分別測(cè)定飽和吸附量。

式中:V0為濕樹(shù)脂體積;c0為上柱液濃度;c1為10%上柱液濃度;V1為當(dāng)流出液濃度為10%上柱液濃度時(shí)的流出液總體積。
1.3.2 動(dòng)態(tài)吸附實(shí)驗(yàn)
將去甲基萬(wàn)古霉素上柱液用水稀釋成濃度為0.8 mg·mL-1、1.6 mg·mL-1、2.2 mg·mL-1、2.8 mg·mL-1的上樣液,以1.5 BV·h-1的流速上樣,考察上樣液濃度對(duì)樹(shù)脂動(dòng)態(tài)飽和吸附量的影響。
分別用5%鹽酸或10%的氫氧化鈉將去甲基萬(wàn)古霉素上柱液 (濃度為2.2 mg·mL-1,pH值為3.5)的pH值調(diào)至3.0、4.0、6.0、7.0、 8.0、9.0、10.0,以1.5 BV·h-1的流速上樣,考察上樣液的pH值對(duì)樹(shù)脂動(dòng)態(tài)飽和吸附量的影響。
將pH值為9.0、濃度為2.2 mg·mL-1的去甲基萬(wàn)古霉素上柱液上HZ816樹(shù)脂柱吸附,上樣流速(BV·h-1)分別為0.5、1、1.5、2、2.5、3和4,考察上樣流速對(duì)樹(shù)脂動(dòng)態(tài)飽和吸附量的影響。
1.4 解吸條件研究
選用甲醇、乙醇、丙酮三種溶劑進(jìn)行靜態(tài)解吸,篩選效果最好的解吸劑。
取一定量的HZ816樹(shù)脂濕法裝入100 mL玻璃樹(shù)脂柱(R∶H=1∶5)中,通上樣液吸附,當(dāng)流出液濃度接近上樣液濃度10%時(shí),認(rèn)為樹(shù)脂已吸附飽和并停止上樣,用純化水洗滌樹(shù)脂后,然后選用5%乙醇-0.1%鹽酸-水、10%乙醇-0.1%鹽酸-水、15%乙醇-0.1%鹽酸-水、20%乙醇-0.1%鹽酸-水、25%乙酸-0.1%鹽酸-水解吸,解吸流速控制在100 mL·h-1,合并高濃度的解吸液,并以HPLC檢測(cè)流出液濃度。
1.5 HPLC色譜條件
色譜條件:色譜柱 Diamonsil C18(4.6 mm×250 mm,5 μm);柱溫為40℃,流動(dòng)相為乙腈∶0.05 mol·L-1KH2PO4=8∶92(體積比),流速1.0 mL·min-1;檢測(cè)波長(zhǎng)280 nm,進(jìn)樣量20 μL。在該色譜條件下去甲基萬(wàn)古霉素的保留時(shí)間為9.5 min。
稱(chēng)取27 mg去甲基萬(wàn)古霉素對(duì)照品配制成50 mL水溶液,取對(duì)照品溶液20 μL注入色譜儀,按上述色譜條件進(jìn)行測(cè)定,按外標(biāo)法計(jì)算峰面積。本方法測(cè)得去甲基萬(wàn)古霉素平均回收率為99.8%(n=5),RSD=0.15%;精密度為99.88%(n=5),RSD=0.20%。
2 結(jié)果與討論
2.1 樹(shù)脂篩選結(jié)果
實(shí)驗(yàn)中測(cè)得8種大孔樹(shù)脂的飽和吸附量,結(jié)果見(jiàn)表1。
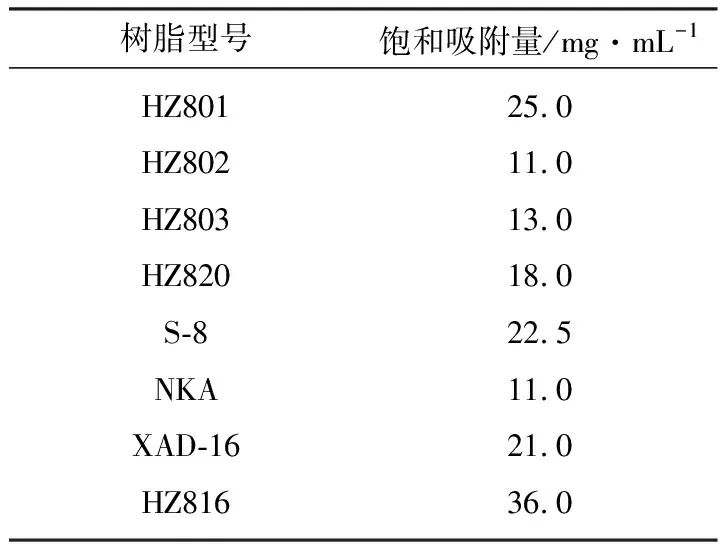
表1 8種大孔樹(shù)脂的飽和吸附量
從表1可見(jiàn),HZ816樹(shù)脂的飽和吸附量最高。因此,選取HZ816型樹(shù)脂作為去甲基萬(wàn)古霉素的吸附樹(shù)脂。
2.2 吸附條件對(duì)HZ816樹(shù)脂動(dòng)態(tài)飽和吸附量的影響
2.2.1 上樣液濃度(圖2)

圖2 上樣液濃度對(duì)HZ816樹(shù)脂動(dòng)態(tài)飽和吸附量的影響
由圖2可以看出,去甲基萬(wàn)古霉素濃度對(duì)HZ816樹(shù)脂動(dòng)態(tài)飽和吸附量有一定的影響。當(dāng)上樣液濃度較低(如0.8 mg·mL-1)時(shí),樹(shù)脂動(dòng)態(tài)飽和吸附量較低,這說(shuō)明仍有部分樹(shù)脂未參與吸附過(guò)程,樹(shù)脂使用率不高;當(dāng)上樣液濃度為2.2 mg·mL-1時(shí),樹(shù)脂動(dòng)態(tài)飽和吸附量最高;而當(dāng)上樣液濃度過(guò)高時(shí),樹(shù)脂動(dòng)態(tài)飽和吸附量也稍有下降,這可能是由于一些雜質(zhì)與萬(wàn)古霉素競(jìng)爭(zhēng)吸附,從而影響了樹(shù)脂對(duì)萬(wàn)古霉素的吸附。選擇較適上樣液濃度為2.2 mg·mL-1。
2.2.2 上樣液pH值(圖3)

圖3 上樣液pH值對(duì)HZ816樹(shù)脂動(dòng)態(tài)飽和吸附量的影響
由圖3可以看出,上樣液pH值為9.0時(shí),HZ816樹(shù)脂對(duì)去甲基萬(wàn)古霉素的動(dòng)態(tài)飽和吸附量較高。這可能是由于去甲基萬(wàn)古霉素呈弱堿性,在弱堿性條件下能夠保持分子狀態(tài),較易被吸附。因此,選擇較適上樣液pH值為9.0。
2.2.3 上樣流速(圖4)

圖4 上樣流速對(duì)HZ816樹(shù)脂動(dòng)態(tài)飽和吸附量的影響
由圖4可以看出,當(dāng)上樣流速超過(guò)2.5 BV·h-1后,樹(shù)脂動(dòng)態(tài)飽和吸附量逐漸下降。流速越慢,吸附越好;但流速過(guò)慢會(huì)延長(zhǎng)生產(chǎn)周期,提高成本。因此,選擇較適的上樣流速為2 BV·h-1。
2.3 解吸條件的確定
2.3.1 解吸溶劑的選擇
解吸實(shí)驗(yàn)結(jié)果表明,乙醇和甲醇均具有較好的解吸能力,因乙醇毒性遠(yuǎn)遠(yuǎn)低于甲醇,故選用乙醇作為吸附樹(shù)脂的解吸溶劑。
2.3.2 解吸劑的確定(表2)
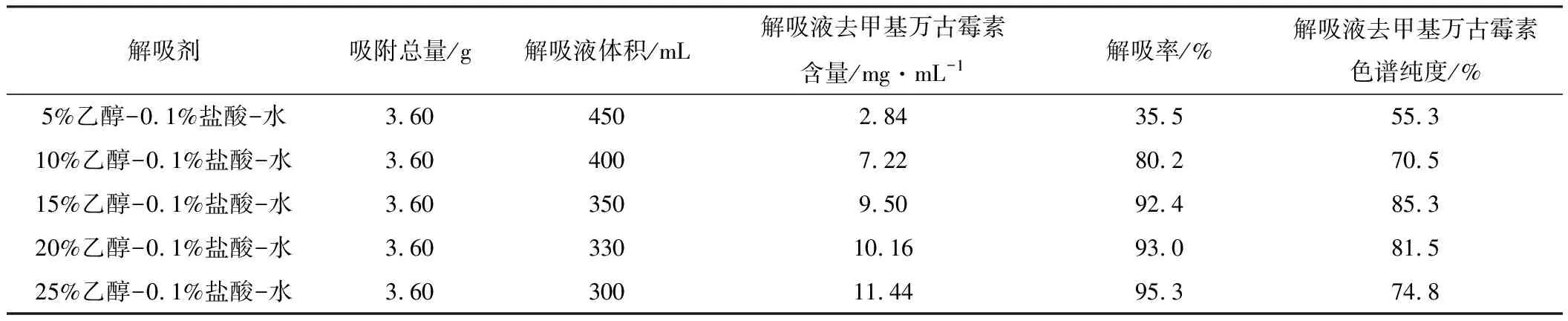
表2 解吸實(shí)驗(yàn)結(jié)果
由表2可知,用較低含量乙醇解吸時(shí)解吸拖尾嚴(yán)重,解吸率低,這可能是由于有機(jī)溶媒含量低時(shí),一方面樹(shù)脂的溶脹度不夠,去甲基萬(wàn)古霉素從內(nèi)孔的溶出受阻,另一方面流動(dòng)相對(duì)去甲基萬(wàn)古霉素的溶解度低,致使其難以進(jìn)入流動(dòng)相而滯留于樹(shù)脂;隨著乙醇含量的上升,解吸率明顯增加,所需解吸液體積減少。用15%乙醇-0.1%鹽酸-水解吸時(shí),解吸液甲基萬(wàn)古霉素的色譜純度最高,表明此時(shí)的雜質(zhì)最少。綜合考慮解吸率和解吸液色譜純度,選取15%乙醇-0.1%鹽酸-水作為解吸劑。
2.4 工藝驗(yàn)證
本工藝經(jīng)過(guò)5批中試放大,結(jié)果表明:大孔吸附樹(shù)脂HZ816可以很好地提取分離去甲基萬(wàn)古霉素,動(dòng)態(tài)飽和吸附量達(dá)36.1 mg·mL-1,可將發(fā)酵液中去甲基萬(wàn)古霉素濃度富集15倍左右;純化水洗樹(shù)脂柱后,采用15%乙醇-0.1%鹽酸-水溶液對(duì)樹(shù)脂柱解吸,解吸液去甲基萬(wàn)古霉素較集中,基本不拖尾,平均解吸率92.0%;去甲基萬(wàn)古霉素色譜純度由發(fā)酵液的低于20%提高到解吸液的85.0%,表明發(fā)酵液中大量色素、蛋白、多糖等雜質(zhì)得以有效去除,為后續(xù)進(jìn)一步純化精制打下了良好的基礎(chǔ)。
3 結(jié)論
選擇HZ816型樹(shù)脂為去甲基萬(wàn)古霉素的吸附樹(shù)脂,在上樣液濃度為2.2 mg·mL-1、pH值為9.0、上樣流速為2 BV·h-1的最佳吸附條件下,HZ816型樹(shù)脂對(duì)去甲基萬(wàn)古霉素的吸附量達(dá)36.1 mg·mL-1;采用15%乙醇-0.1%鹽酸-水即可將吸附在樹(shù)脂柱上的去甲基萬(wàn)古霉素有效解吸,解吸率達(dá)92.0%。該工藝簡(jiǎn)單、易操作,適用于工業(yè)化生產(chǎn)。
參考文獻(xiàn):
[1] 金有豫.藥理學(xué)[M].北京:人民衛(wèi)生出版社,2001:333-335.
[2] 陳代杰,李繼安,鄒韻華,等.萬(wàn)古霉素的研究開(kāi)發(fā)[J].中國(guó)抗生素雜志,2004,29(1):8-10.
[3] 哈羅德·蒙·卡特,哈羅德·伯納德·海斯.改進(jìn)的萬(wàn)古霉素沉淀法[P].CN 10 212 280 C,1993-06-16.
[4] Grahek Rok,Bastarda Andrej.Chromatographic purification of va-ncomycin hydrochloride by use of preparative HPLC[P].USP 5 854 390 A,1998-12-29.
[5] 郭立安,朱寶泉,陳代杰.萬(wàn)古霉素的磁性親和吸附分離[J].生物工程學(xué)報(bào),2001,17(5):584-586.
[6] 張巧,張奇,楊藝虹.雙水相萃取技術(shù)應(yīng)用在醫(yī)藥工業(yè)中的展望[J].醫(yī)藥工程設(shè)計(jì)雜志,2001,22(5):22-26.
[7] 段金友,方積年.反膠束萃取分離生物分子及相關(guān)領(lǐng)域的研究進(jìn)展[J].分析化學(xué),2002,30(3):365-371.
[8] Pflaum Zlatko,Turkalj Kobert.Combined process for the purification of vancomycin hydrochloride[P].USP 5 853 720 A,1998-12-29.
