化探樣品中微量元素 Ag、W、Mo、Sn、Bi、Cu、Pb、Zn、Ni、Co、Cr的水平撒料光譜定量分析
2010-06-06 12:22:12胡道榮
時代農機 2010年7期
關鍵詞:分析
胡道榮
(中南地質勘查院新疆分院,新疆 烏魯木齊 830063)
地球化學找礦,要求測定化探樣品中痕量元素的檢出下限達到10-6甚至更低的含量。這類樣品通常是大批量的,而且是多元素分析,因此,要求分析方法具有靈敏度高、準確、快速、成本低等特性。
我們所目前分析化探樣品所采用的方法有化學法、原子吸收法、極譜法等,雖然這些方法對W、Mo、Sn、Bi等元素可達到所要求的靈敏度和準確度,但是這些方法測定往往是單元素分別進行,所以不但成本高,而且速度慢,分析周期長。再者,Ag在我所現有原子吸收分光光度計上采用火焰法測定的下限只能達到5×10-6,與化探樣品中Ag的檢出限要求相關甚遠,無法滿足化探樣品的分析。為填補這一空白,根據水平電極撒料法具有快速,靈敏,分餾效應小,對易揮發元素、中等揮發元素甚至某些難揮發元素可以同時測定的特點[1],借鑒其它單位的經驗,對原有的W-100平面光柵攝譜儀進行改裝,我們在前人工作的基礎上做了一些條件試驗,并結合本單位的實際情況,擬定了一個區域化探急需的,要求分析靈敏度較高的準確性要符合定量要求的方法。本法采用了特制的水平電極撒樣漏斗,改善了下料的均勻性,提高了分析精度;同時選用了SiO2、石墨粉及Na2SO4混合緩沖劑,它能提高弧燒的穩定性,使樣品從電極間平緩地進入弧焰,在蒸氣云中保持足夠的原子及離子濃度,獲得了較高的再現性。本法操作簡便、快速、成本低,各元素的檢出限分別為:Ag 0.03×10-6,W 0.5×10-6,Mo 0.05×10-6,Sn 0.4 ×10-6,Pb 1×10-6,Zn 3×10-6,Cu 0.5×10-6,Bi 0.1×10-6,Ni 0.3×10-6,Co 0.3×10-6,Cr 1×10-6,基本上達到了化探普查找礦定量分析的要求。
1 標準配制
(1)人工基物組分:SiO270%;Al2O315%;Fe2O35%;CaCO32%;MgO 2%;K2SO43%;Na2SO43%。
(2)標準系列:準確稱取定量的 Ag、W、Mo、Sn、Bi、Cu、Pb、Zn、Ni、Co、Cr的光譜純氧化物加入上述人工基物(A)中,使基體中金屬含量 Cu、Pb、Zn、W、Sn、Bi、Co、Ni、Cr為 0.3%;Ag、Mo為0.03%,研磨均勻后即得地(1)號標準。
即:(1)號:0.3%Cu、Pb、Zn、W、Sn、Bi、Co、Ni、Cr;0.03%Mo、Ag。
然后按1、3級差用人工基物(A)沖稀到(9)號,得到一套標準系列,詳見表1。

表1 標準系列
2 儀器及工作條件
2.1 攝譜條件
攝譜儀:W-100一米平面光柵攝譜儀;光柵刻線1200條/mm,閃耀波長570 nm,二級光譜;狹縫7 μ;三透鏡照明系統;中間光欄5 mm×10 mm。
激發光源:WPF-2交流電弧發生器,輸入電壓220 V,電流強度20 A。
預燃時間:5 s。
曝光時間:16 s(I=20A),下料 300 mg。
電極:直徑6 mm左右電極均為錐形頭。
感光板:天津I型紫外感光板。
2.2 暗室處理及光度測量
暗室處理:按國產板A、B液及定影液配方配制顯影液和定影液,在20℃顯影3 min,于定影液中定影至透明,約5~10 min,水沖洗 10 min。
光度測量:東德GFE760 μ測微光度計,P標尺,測量狹縫寬26 μ,高16 mm。
3 水平電極撒料裝置的制備
與垂直電極裝樣法相比,水平電極撒樣法除省去車制電極的繁瑣外還有著不少的優點[2]:它在燃弧時間內,能保證大量試樣均勻地進入弧焰,發射譜線強度大,因而能改善元素的檢出限,同時可縮短曝光時間,提高分析速度;此外,它的弧焰較為穩定,且分餾效應小,因此,特別適合于化探樣品中的多元素分析。
目前,撒樣法最廣泛和普遍采用的下樣方法是滑槽或傳送帶下樣,滑槽法樣品下到電弧中的速度不易控制,而傳送帶法容易引起樣品沾污,而且這兩種方法進入弧焰中的樣品都不夠均勻,再加上下料漏斗口都較大,一般為3~4 mm(如太小則易發生堵塞),這樣由于樣品粒度、粘性及其他因素影響造成試樣沿漏斗壁不同位置下到電弧中的位置便會相應變化而引入偶然誤差。對此我們采用了一種特制漏斗,如圖1所示。這種漏斗主要是通過振蕩器的振蕩,使得漏斗上的玻璃棒產生振動及轉動,從而使漏斗中的樣品能夠以一定的速度均勻地進入弧焰,因此它大大改善了下料的均勻性。
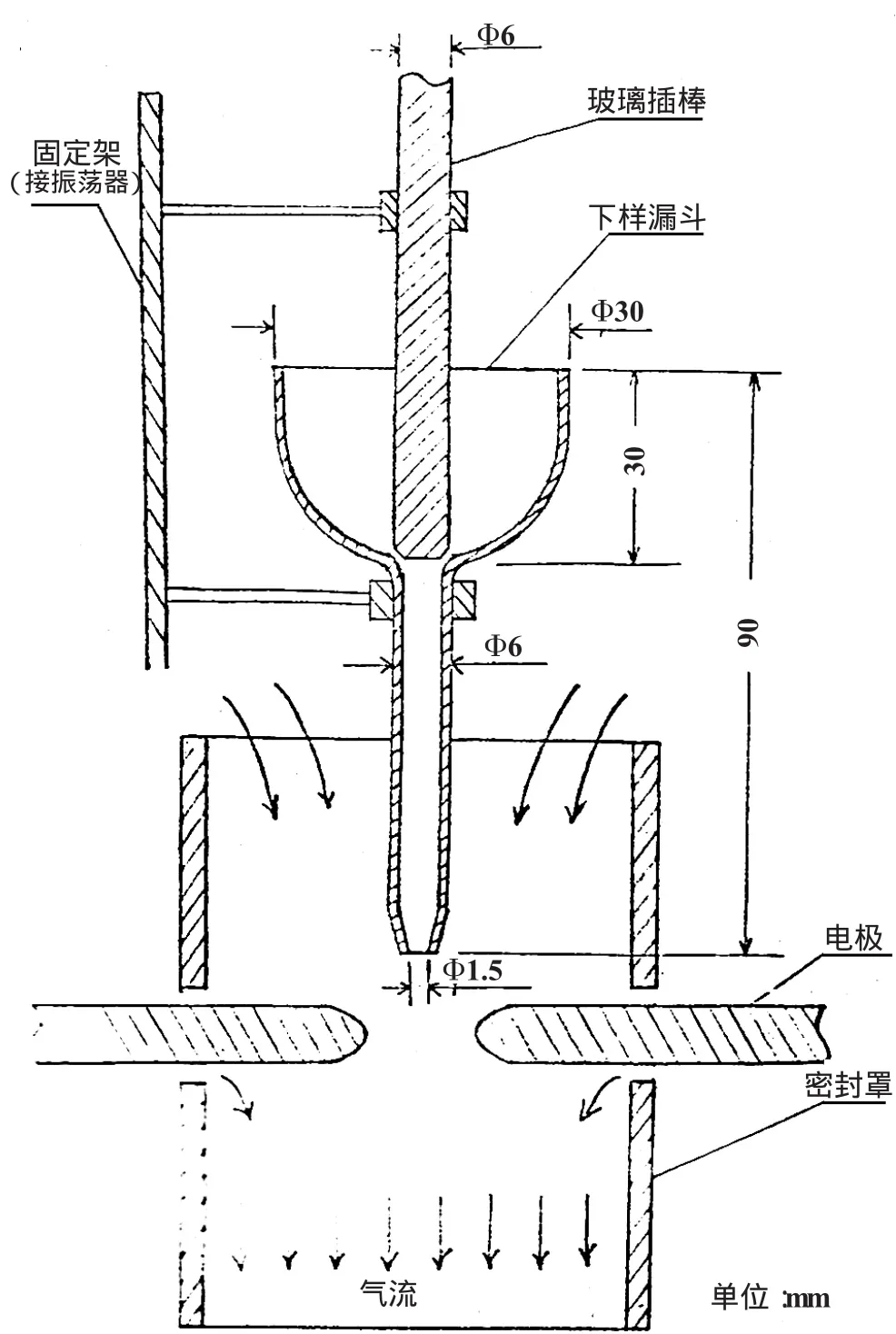
圖1 水平電極撒樣裝置
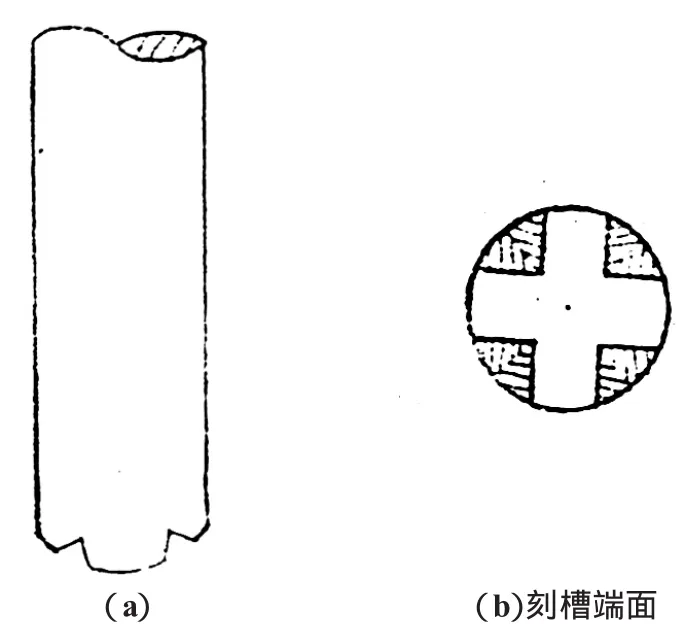
圖2 四刻槽平頭形插棒
我們發現玻璃插棒頭的形狀對下料的速度、均勻性及樣品沾污程度的影響較大。我們對三角錐形、四角錐形、三槽三角錐形、三槽四角錐形、四槽園錐形、三槽平頭形、四槽平頭形等不同形狀的插棒頭進行了對比試驗,發現四槽平頭形插棒的效果最佳,這種插棒的下料比較均勻,而且還能減少沾污,因此采用了如圖2所示的四刻槽平頭形插棒。
4 單元素線減光器的制作及應用
本法中的Ag的分析線選擇3280.7A和3382.9A,其中,3280.7的測定上限為3×10-6,而3382.9A的測定上限也只能測到5×10-6,為了使Ag5×10-6~30×10-6可以測定而又不降低其他元素分析線的靈敏度,我們采用了單元素、單譜線的減光器,將它固定于暗箱出口處Ag3382.9A譜線的相應位置上,結果證明達到預期目的。
這種減光器的制作:選擇透明度好,厚度約0.1 mm的云母片,擦洗干凈后放入真空鍍膜機去鍍一薄層Au薄。其鍍膜工藝是[4]:將10~15 mg的金絲或Au薄掛在鍍膜機的W絲上,加熱使Au熔化沾在W絲上后,停止加熱,抽真空,達到真空度后加熱噴鍍,取出后剪成3×12(mm×mm)的鍍Au云母片,貼在做好的架上,置于光譜出口位置上。如圖3所示。
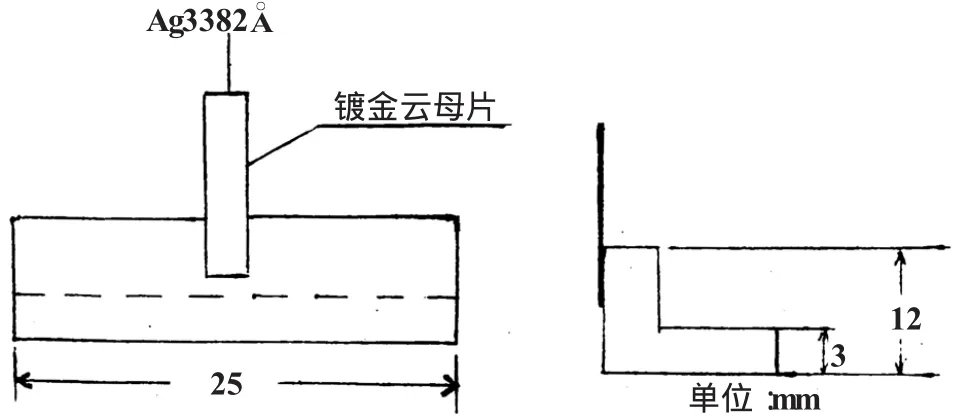
圖3
5 內標元素的選擇
在光譜定量分析中,為了克服工作條件變化對譜線強度帶來的影響,常需引入內標元素。
由于撒樣法的各元素蒸發特性曲線目前條件下無法實驗和繪制,所以內標的選擇只能從查閱資料著手,參考垂直電極法的蒸發曲線,結合嘗試法來實現。為此我們對易揮發元素選擇Ge、In、Sb;對中等揮發及難揮發元素選擇Sc、Au、Pd作比較,實驗表明,Ge和Pd的效果較為理想,因此選用Ge作Pb、Zn、Ag、Bi、Sn、Mo的內標,用 Pd作 Cu、Ni、Co、W、Cr的內標。
6 緩沖劑的選擇及配制
緩沖劑的作用,在多元素分析中很重要,特別是化探樣品組分變化復雜,當標準與樣品組分不一致時,則產生標準元素黑度值大,樣品分析元素黑度值小,使結果偏低的影響,反之則會使結果偏高。因此,在光譜定量分析中往往引入緩沖劑,其目的主要有:消除組分影響;提高被測元素的靈敏度;改善分析方法的再現性等。
由于低電離電位元素(如K、Na等堿金屬元素)對電弧溫度影響很大,而所分析樣品中K和Na的量變化較大,如果在緩沖劑中不加入一定量低電離電位元素,就必然影響測定的穩定性和準確性。據資料[3]介紹,K2SO4能穩定弧焰,改善弧燒狀況。而資料[4]又報導,若在緩沖劑中加入NaF,則電弧溫度將隨NaF量的變化而變化,而當緩沖劑中NaF量為2%~6%時弧溫變化較小,為此我們選擇了含低電離電位元素的化合物(如KCl、KClO3、NaF、Na2SO4、K2SO4等)作穩定劑,以 SiO2,基體及 C粉等作攜帶劑和稀釋劑,將它們按不同的組合及不同的比例混合,進行試驗比較,發現SiO2、C粉中加NaF與SiO2、C粉中加Na2SO4的效果都不錯,而且后者效果更好。最后,選擇了SiO2∶C∶Na2SO4=61∶30∶9 的混合物作緩沖劑。這種緩沖劑能提高弧燒的穩定性,使樣品從電極間平緩地進入弧焰,在蒸氣云中保持足夠的原子及離子濃度,獲得了較高的再現性,同時能使W、Mo、Bi等元素的分析靈敏度稍有提高。
在配制緩沖劑時還加入了內標元素,其組成為:SiO261 g;C(石墨粉)30g;Na2SO49g;GeO20.125g;PdO0.006g。將它們混合磨勻備用。
7 分析手續及分析線對
按樣品∶緩沖劑=1∶1的混合比例,分別稱取樣品(包括被測樣品、標準樣品、管理樣品)150 mg和緩沖劑150 mg,混合磨勻備用。
在前面所述的儀器及工作條件下進行攝譜、洗板和測光,以△P-logC繪制工作曲線,查出含量。
測定所采用的分析線對見表2。
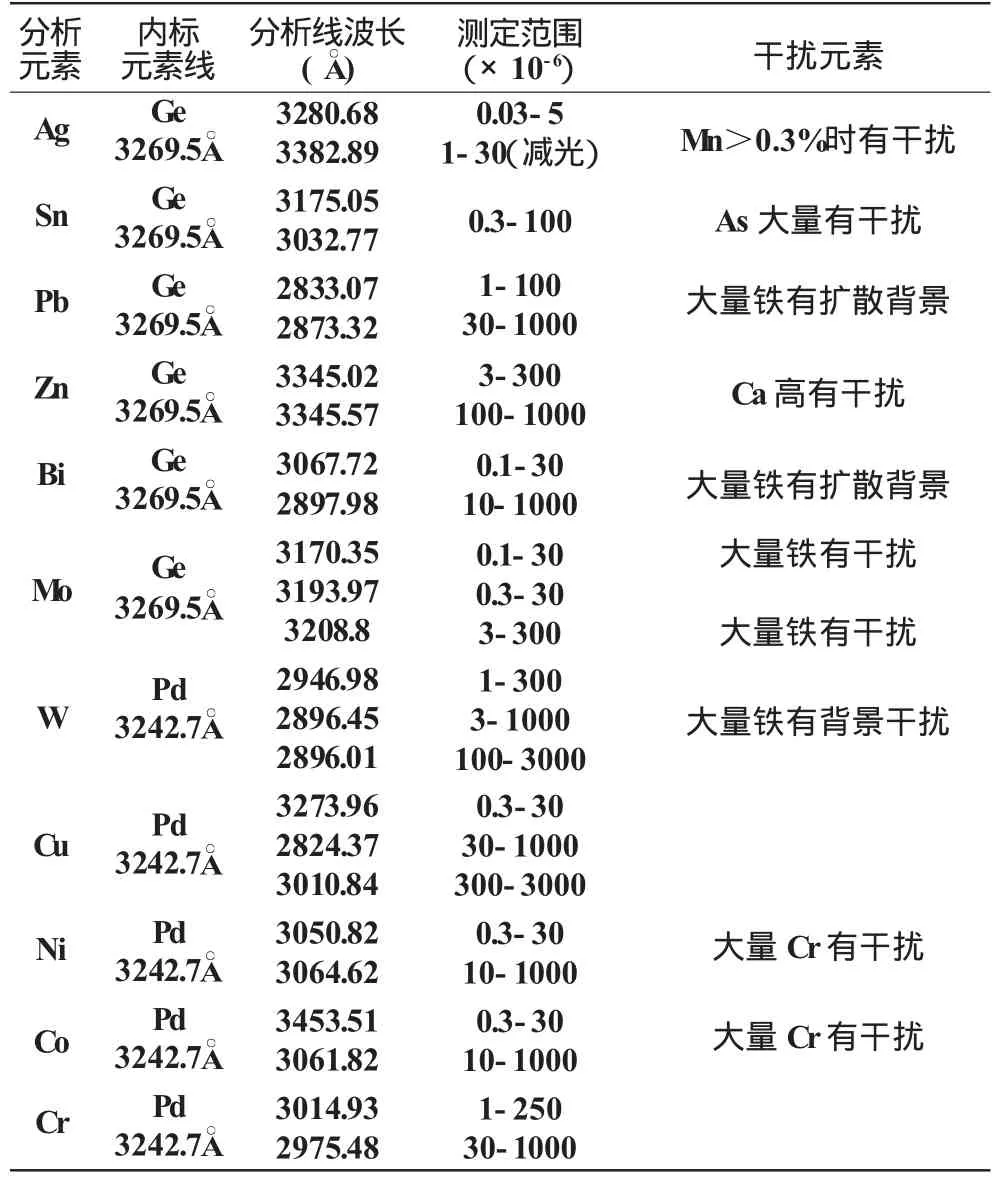
表2 分析線對及測定范圍
8 方法的檢出限和精密度
8.1 檢出限
檢出限是衡量分析方法優劣的重要技術指標之一,它通常是指某一分析方法能可靠地檢出試樣中某元素的最低值。為此,我們取空白樣品攝譜20次,按檢出限公式:XL=+KS0進行測量計算求出。式中為空白樣品測量信號值的平均值,S0為空白樣品測量信號值的標準偏差,K為根據選定的置信度所確定的常數值,當K=3時,XL為元素的分析線的檢出限信號值,然后以此在標準工作曲線或其延長線上查出的CL值,即為所測元素分析線的檢出限。本法測得的各元素的檢出限列于表3。
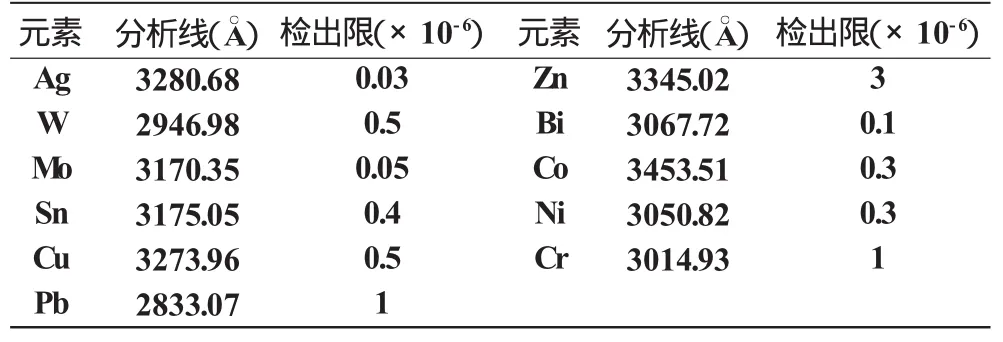
表3 各元素的檢出限
8.2 精密度
我們選用了四個綜合樣品各進行15次攝譜分析,得出各元素的每次含量及15次的平均含量,然后計算出各元素在某含量時的相對標準偏差(RSD%),即為本方法的精密度。見表4。
9 回收試驗
在人工配制的基體中加入一定量的被測元素,然后按本法的工作條件進行測定分析,求出含量,并計算回收率。見表5。
10 標樣分析結果及對照
用本法對十個地球化學標準樣品(GSD、MGD)進行三次分析,各元素分析結果的平均值與標樣定值的對照情況列于表6。
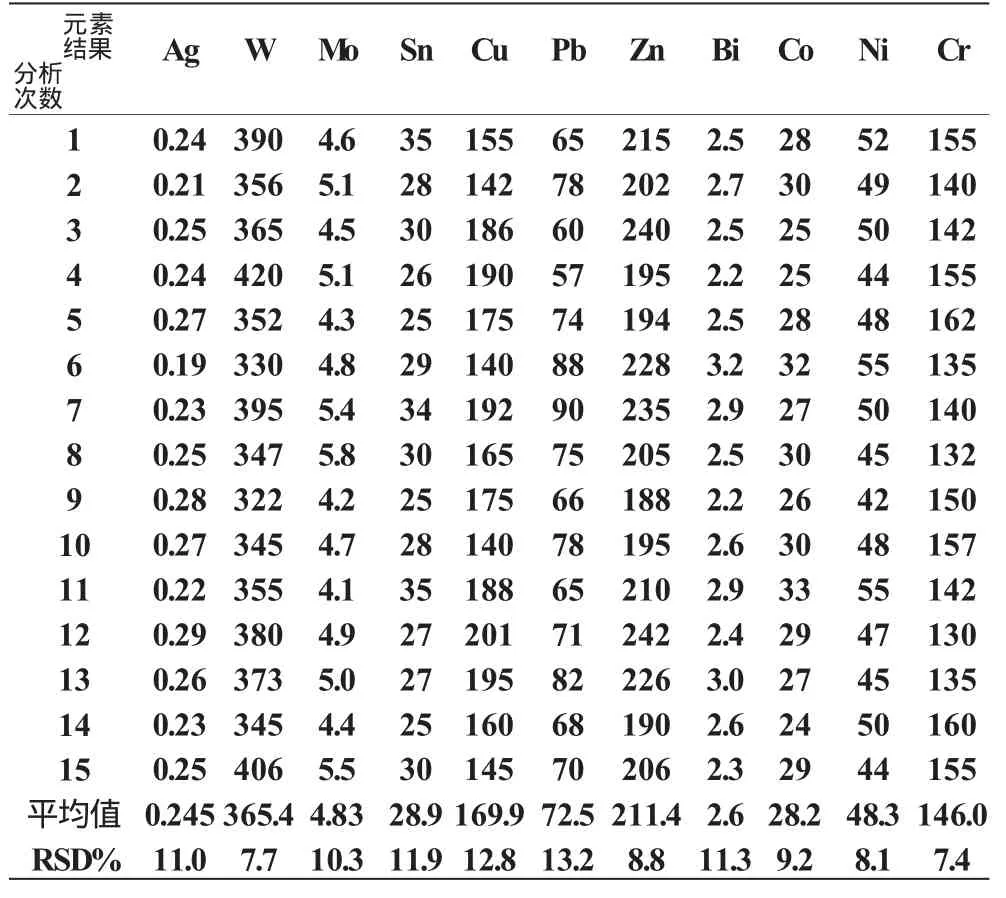
表4 精密度(分析結果單位:W(B)/10-6)

表6 GSD、MGD分析單位:W(B)/10-6
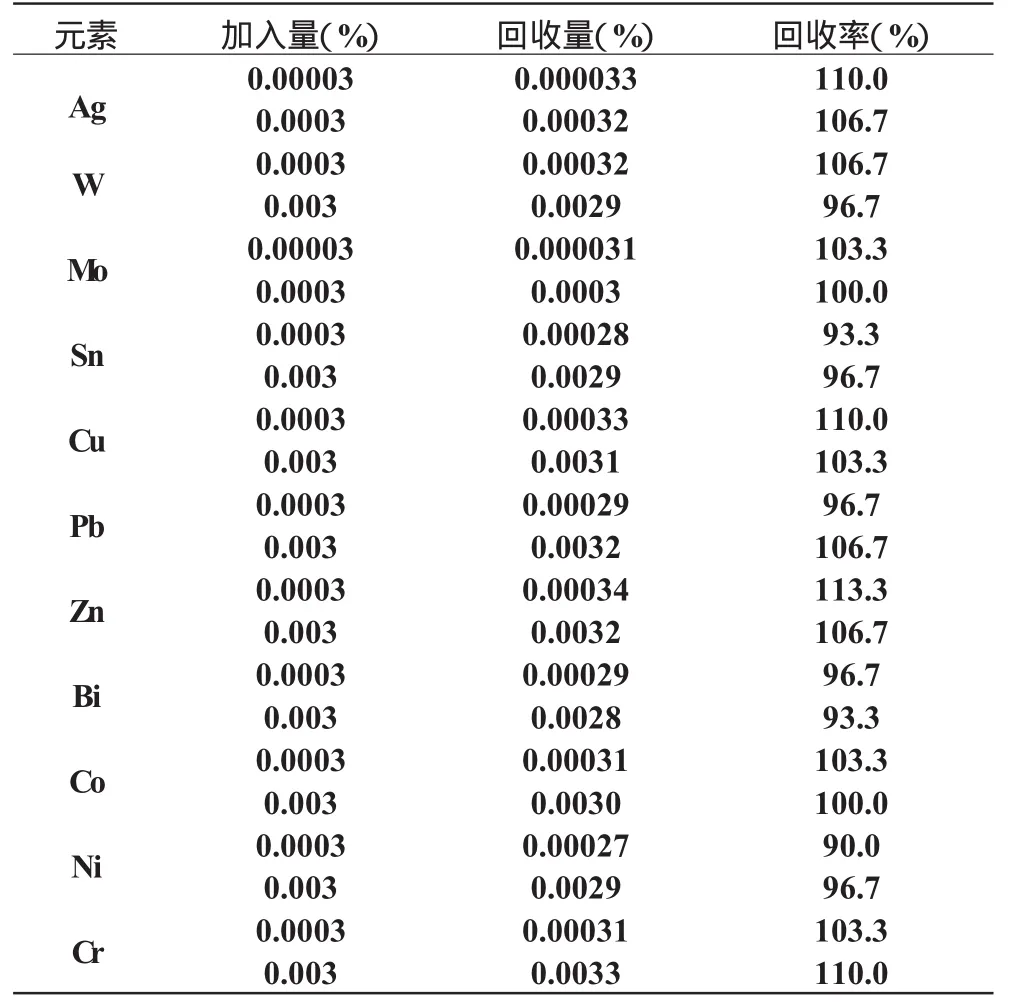
表5 回收率
11 討論
(1)本法適用于以硅酸鹽為主的樣品分析,樣品中大量的鐵、鈣等會對 Pb、Zn、Bi、W、Mo等元素產生干擾。
(2)本法靈敏度較高,所分析元素的檢出限基本達到或超過中國有色金屬總公司地質局九二年下發的《有色地質分析測試質量管理辦法》的要求。
(3)本法省去了車制電極的繁瑣,操作比較簡單,成本較低。
(4)為校正工作曲線,需攝較多的管理樣品。
(5)實驗表明此法再現性較好,但條件變化引起的誤差較大,只要嚴格控制條件是可以保證精度的。對于化探樣品,如操作比較熟練,每個樣品只攝一條譜就可以了,對地質樣品或不熟練的操作者,可以采取稱樣品0.3 g和緩沖劑0.3 g,混勻后分兩次攝兩條譜進行測定。
(6)采用瑪瑙乳缽混樣,即費時又易引起樣品間的玷污,為此,我們將稱好的樣品與緩沖劑放入特制托盤中的2~3 mL瓷坩堝內,鋪上干凈白紙,密閉蓋嚴后,置于振蕩器中振蕩混勻,這樣即省時又避免了玷污。另外,為使下樣時不易發生沾堵,最好將混好的樣品于烘箱中烘干后攝譜或于干燥器中保存備用。
[1]單永健.南嶺鎢礦賦存層位中的微量 W、Sn、Bi、Mo、Be、Cu、Sb 光譜定量測定[J].地質地球化學,1982,(1).
[2]李廷均.發射光譜分析[M].北京:原子能出版社,1983.
[3]劉詩達.化探樣品中微量Sn、Bi、Cd的光譜定量測定,1985.
猜你喜歡
現代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
當代經濟研究(2016年5期)2016-12-01 03:12:05
現代農業(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學學報(社會科學版)(2014年3期)2014-04-16 04:38:31
終身教育研究(2014年5期)2014-02-28 01:23:06
