
酸水解條件對大黃及其制劑中蒽醌類成分含量的影響研究
2010-09-17 01:32:18封淑華韓桂茹
中成藥 2010年7期
封淑華, 韓桂茹, 馮 麗
(河北省藥品檢驗所,河北石家莊050011)
大黃是一種臨床上應(yīng)用最廣泛的中藥,主含蒽醌類成分,以蒽醌苷及其苷元存在于植物體內(nèi)。藥理研究表明:游離大黃蒽醌具抗菌、消炎、止血等作用;結(jié)合大黃蒽醌具較強的瀉下作用[1]。在中藥成方制劑中有采用大黃瀉下功效的[2],有采用其抗菌、消炎、止血功效的。其質(zhì)量控制方法目前為止都是測定的水解后的大黃總蒽醌,該指標無法區(qū)分制劑的瀉下和抗菌、消炎功效。筆者試采用同一流動相,分別測定制劑中的總蒽醌、游離蒽醌和結(jié)合蒽醌的含量,按功效分別控制制劑的結(jié)合與游離蒽醌含量,實現(xiàn)了測定指標與功效結(jié)合的愿望。但在測定過程中發(fā)現(xiàn)同一樣品,游離大黃酸的含量大于總大黃酸含量。對此,進行探討研究,判明了發(fā)生的原因。報道如下:
1 儀器與試藥
Waters2695高效液相色譜儀(美國 Waters公司)。
大黃素(批號:110756—200110)、大黃酚(批號:110796—200513)、大黃酸 (批號:110757—200206)、大黃素甲醚(批號:110758—200509)、蘆薈大黃素(批號:110795—200504),購自中國藥品生物制品檢定所;三黃片(批號:080202)由河南宛西制藥廠提供;麻仁潤腸丸(批號:7013195)由北京同仁堂藥廠提供。乙腈、甲醇、磷酸為色譜純,其它試劑均為分析純。
2 方法與結(jié)果
2.1 色譜條件 Shimadzu C18色譜柱(4.0 mm×250 mm,4.5 μm),流動相為乙腈-甲醇-0.1%磷酸(42∶23∶35),流速1.0 mL/min。檢測波長254 nm。
2.2 對照品溶液的制備 精密稱取大黃素、大黃酚、大黃酸、大黃素甲醚、蘆薈大黃素適量,分別加甲醇溶解制成濃度為每1 mL含5~10μg的溶液,搖勻,即得。
2.3 供試品溶液的制備
2.3.1 總蒽醌溶液的制備 精密稱取大黃藥材粉末0.3 g(或三黃片粉末0.5 g、麻仁潤腸丸小碎粒1 g),分別精密加入不同比例的甲醇-鹽酸溶液25 mL,稱重,麻仁潤腸丸浸泡10 h以上,超聲使溶散,置80℃水浴中回流30 min,放冷至室溫,補足重量,搖勻,濾過,精密量取續(xù)濾液1 mL(麻仁潤腸丸2 mL),至10 mL(麻仁潤腸丸5 mL)量瓶中,加2%的氫氧化鈉溶液0.5~1 mL,加甲醇定容至刻度,搖勻,濾過,取續(xù)濾液作為測定總蒽醌的供試品溶液。
2.3.2 游離蒽醌溶液的制備 精密稱取大黃藥材粉末0.1 g(或三黃片粉末0.3 g、麻仁潤腸丸小碎粒0.8 g),精密加甲醇25 mL,麻仁潤腸丸浸泡10 h以上,用玻棒研磨使樣品溶散,用數(shù)滴甲醇沖洗玻棒于錐形瓶中,超聲處理(功率160 W,頻率50 kHz)30 min,放冷,再稱定重量,用甲醇補足或揮散至原重量,搖勻,濾過,取續(xù)濾液作為測定游離蒽醌的供試品溶液。
2.4 測定 精密量取對照品溶液和供試品溶液各5~10μL,注入液相色譜儀,記錄色譜圖,色譜峰面積。對照品溶液和大黃藥材溶液總蒽醌色譜圖、游離蒽醌色譜圖見圖1~圖3;不同酸濃度下的三黃片總蒽醌和游離蒽醌各峰面積見表1、各成分的變化趨勢,見圖4、5、6;不同酸濃度下的麻仁潤腸丸總蒽醌和游離蒽醌各峰面積見表2、各成分的變化趨勢,見圖 7、8。

圖1 大黃5種對照品HPLC色譜圖

圖2 大黃藥材中總大黃蒽醌HPLC色譜圖

圖3 大黃藥材中游離大黃蒽醌HPLC色譜圖
從圖1~圖3,清楚看到大黃中5種蒽醌成分分離良好,總大黃蒽醌樣品中的波峰數(shù)多于游離大黃蒽醌樣品中波峰數(shù),而且總大黃蒽醌中的波峰2與波峰6,在不同酸度下發(fā)生相互轉(zhuǎn)變,即波峰2隨著酸濃度的減小,峰面積升高,波峰6隨著酸濃度的減小,峰面積在縮小,且不同酸濃度下的波峰2和波峰6面積相加,近于一固定值。
3 討論
3.1 從圖2、3得知,總大黃蒽醌中的波峰2(大黃酸)與波峰6,在不同酸度下發(fā)生相互轉(zhuǎn)變,二峰面積之和基本是一定值,5個已知組分可以準確計算含量,而波峰6成分未知,無法計算含量,在控制稱樣量幾乎一致的情況下,選擇以峰面積表示其相對變化趨勢更為直觀。

表1 不同酸解條件下三黃片中各蒽醌成分的峰面積值

圖4 不同酸濃度下三黃片中蘆薈大黃素、大黃素、大黃酚、大黃素甲醚的峰面積變化趨勢圖

圖5 不同酸濃度下三黃片中大黃酸、波峰6的峰面積變化趨勢圖
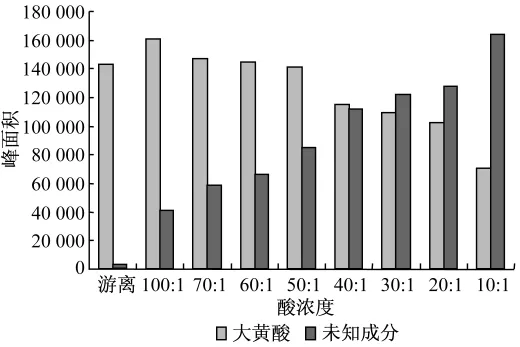
圖6 不同酸濃度下三黃片中大黃酸、波峰6的峰面積變化趨勢柱形圖

圖7 不同酸濃度下麻仁潤腸丸中蘆薈大黃素、大黃素、大黃酚、大黃素甲醚的峰面積變化趨勢圖
3.2 通過上述探討研究得知,大黃或含大黃的制劑在同時測定總大黃蒽醌與游離大黃蒽醌時,水解酸度對蘆薈大黃素、大黃素、大黃酚、大黃素甲醚的波峰面積幾乎無影響,也就是酸度大小不影響上述四成分的含量,但對大黃酸的影響卻比較嚴重。從表1的數(shù)值看到要使三黃片中總大黃酸大于游離大黃酸,甲醇-鹽酸的濃度要在60∶1~100∶1之間;從表2的數(shù)值看到,麻仁潤腸丸中,即便甲醇∶鹽酸的濃度減小到100∶1,總大黃酸的濃度仍然比游離的低,是一負值。提示由于組方不同,影響因素而不一樣,如要以大黃酸為指標進行含量測定,必須具體品種具體考察酸水解濃度。
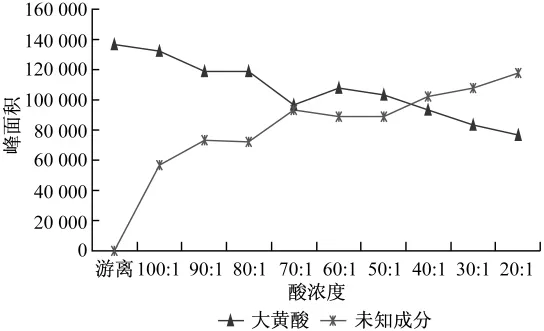
圖8 不同酸濃度下麻仁潤腸丸中大黃酸、波峰6的峰面積變化趨勢柱形圖
3.3 從表1、2的測定數(shù)值還了解到,在不同酸度下三黃片和麻仁潤腸丸中的大黃酸都是隨著酸濃度的減小,峰面積增大,同時與其相對應(yīng)的未知峰6,則隨著酸濃度的減小而減小,二峰面積之和基本是一定值,三黃片中二峰之和在20~23萬之間變化(因取樣量稍有差異);麻仁潤腸丸中二峰之和在18~19萬之間變化。數(shù)據(jù)提示:大黃酸隨著酸度升高,進一步水解為極性小的成分。為確定其是否同一母核化合物,對大黃酸和波峰6,采用二極管陣列檢測器,進行了光譜掃描,結(jié)果證明二峰為同一母核化合物,大黃酸進一步分解為波峰6。光譜掃描見圖9。

表2 不同酸解條件下麻仁潤腸丸中各蒽醌成分的峰面積值

圖9 大黃酸與未知成分的光譜掃描圖
3.4 通過對中藥代表性片劑和蜜丸的酸水解濃度考察,發(fā)現(xiàn)三黃片隨著酸濃度的減小,水解后的樣品過濾速度減慢,到甲醇-鹽酸為100∶1時,幾乎無法過濾;但麻仁潤腸丸則無此情況產(chǎn)生,分析是片劑中的輔料影響了過濾速度。
3.5 從被測定成分的保留和運行時間、波峰面積大小、分離情況以及影響因素等綜合考慮,認為在中藥成方制劑中如若以大黃為含量測定目標藥材,以大黃素和大黃酚為測定指標比較適宜。二者波峰面積占5種成分面積之和的約60% ~70%,而且不受酸水解濃度影響,運行時間可調(diào)整在20~30 min之內(nèi),大黃素、大黃酚和大黃素甲醚波峰分離良好。
3.6 目前各類國家標準制劑中的大黃均是以大黃素、大黃酚為指標進行含量測定的[2],上述研究證實了其測定指標的合理性。
[1]喬傳卓,張漢明,宓鶴鳴.生藥學[M].上海:同濟大學出版社,1995:184-186.
[2]中國藥典[S].一部,2005:604-605.
