阿片受體顯像劑11C-Carfentanil的前體合成及放射性標記
2011-01-09 04:52:02張錦明徐志紅張曉軍向曉輝田嘉禾
核化學與放射化學 2011年4期
關鍵詞:產品
張錦明,桂 媛,徐志紅,張曉軍,向曉輝,田嘉禾
1.中國人民解放軍總醫院核醫學科,北京 100853;
2.江蘇常熟華益化工有限公司,江蘇常熟 215522;3.北京大學神經科學研究所,北京 100191
阿片受體顯像劑11C-Carfentanil的前體合成及放射性標記
張錦明1,桂 媛2,徐志紅2,張曉軍1,向曉輝3,田嘉禾1
1.中國人民解放軍總醫院核醫學科,北京 100853;
2.江蘇常熟華益化工有限公司,江蘇常熟 215522;3.北京大學神經科學研究所,北京 100191
為合成μ-阿片受體顯像劑:1-(2-苯乙基)-4-(N-苯基丙酰胺基)哌啶-4-羧酸[11C]甲酯([11C]-methyl 1-phenethyl-4-(N-phenylpropanamido)piperidine-4-carboxylate,11C-CFN),合成了標記前體:1-苯乙基-4-(N-苯基丙酰胺基)哌啶-4-羧酸,并用11C標記。標記前體和各步合成中間體均經核磁和質譜確證。采用11CTriflate-CH3標記,Sep-Pak柱色層純化,標記率為(67.3±2.3)%(n=3),產物放化純度大于98%,比活度為1.67×1014Bq/g(66.6 PBq/mol),基本滿足臨床研究的需求。
11C;阿片受體;11C-Carfentanil;PET
11C標記的卡分太尼(11C-Carfentanil,11C-CFN)是一個選擇性很強的μ-阿片受體顯像劑,可用于人中樞神經μ-阿片受體的顯像,以研究阿片受體的藥理機制、針炙止痛的機理[1-2]。同時11C-CFN可用于研究癲癇、肺腺癌和煙酒等濫用的監測[3]。Dannals等[4]最先報道了合成11C-CFN,采用11CH3I與去甲基的鈉鹽前體加熱反應,經 HPLC純化,合成時間為30 min。Jewett[5]選用活性更高的11C-CH3-Triflate與前體在四丁基胺堿性條件下反應,采用固相萃取法,結合陰離子交換膜除放射性雜質,合成時間減小到10 min。Shao等[6]采用商業化模塊自動化合成了11C-CFN,但合成效率只有40%,需進一步研究合成并提高其產率。國內最早合成了用于醫療的CFN,但采用CFN直接脫甲基合成CFN前體的效率低。本研究參照朱國政及Wadsak的合成方法[7-8],擬自行合成CFN的前體和標準品,采用11C-CH3-Triflate堿性條件下標記11C-CFN,改良的柱色層法純化標記藥物,以期達到較高的合成效率。
1 試劑和儀器
N-(2-苯乙基)-4-哌啶酮、氰化鉀、苯胺、濃硫酸、氫氧化鉀、碘甲烷、丙酸酐、六次甲基磷酸三酰胺、Pd/C等購自上海阿拉丁試劑有限公司;冰醋酸購自國藥試劑公司。所有試劑均為分析純,購買后沒有純化直接使用。低本底1 mol/L氫化鋰鋁/THF,德國 ABX公司;DMSO,美國 Alfa Aesar公司;57%的氫碘酸、1 mol/L四丁基胺甲醇溶液,美國 Sigma-Aldrich公司;無水乙醇,美國Millennium公司;氨水,北京化工廠;Sep-Pak C-2柱 ,美國Alltech公司產品;GraceAlltimaC-18分析柱,5μm,10 mm×250 mm,美國 Grace公司。
Agilent 6120型質譜儀,美國安捷倫公司;Bruker 300M核磁共振儀,美國布魯克公司;SGWX-4熔點儀,上海精密儀器廠;Sumitomo HM-20S回旋加速器,日本駐友公司;C-11碘代甲烷合成器,北京派特生物技術有限公司;高效 HPLC分析儀,美國 Waters公司,配515泵、2487紫外檢測器、BioScan流動放射性檢測器。API2000型LC/MS/MS,美國API公司。
2 實驗方法
2.1 去甲基CFN和CFN標準品的合成
去甲基CFN和CFN標準品合成路線[7-8]示于圖1。
2.1.1 4-哌啶乙腈 ,4-苯胺-1-(2-苯乙基)(1)的合成 將 N-(2-苯乙基)-4-哌啶酮6.7 g和苯胺3.7 g溶于24.5 g冰醋酸中,室溫攪拌下加入2.3 g KCN與2.5 g水的溶液,反應48 h后將反應混合物倒入水中,加氨水使其呈堿性,濾出所得固體,用異丙醇重結晶得固體5.7 g(產率55%),熔點118~123℃。
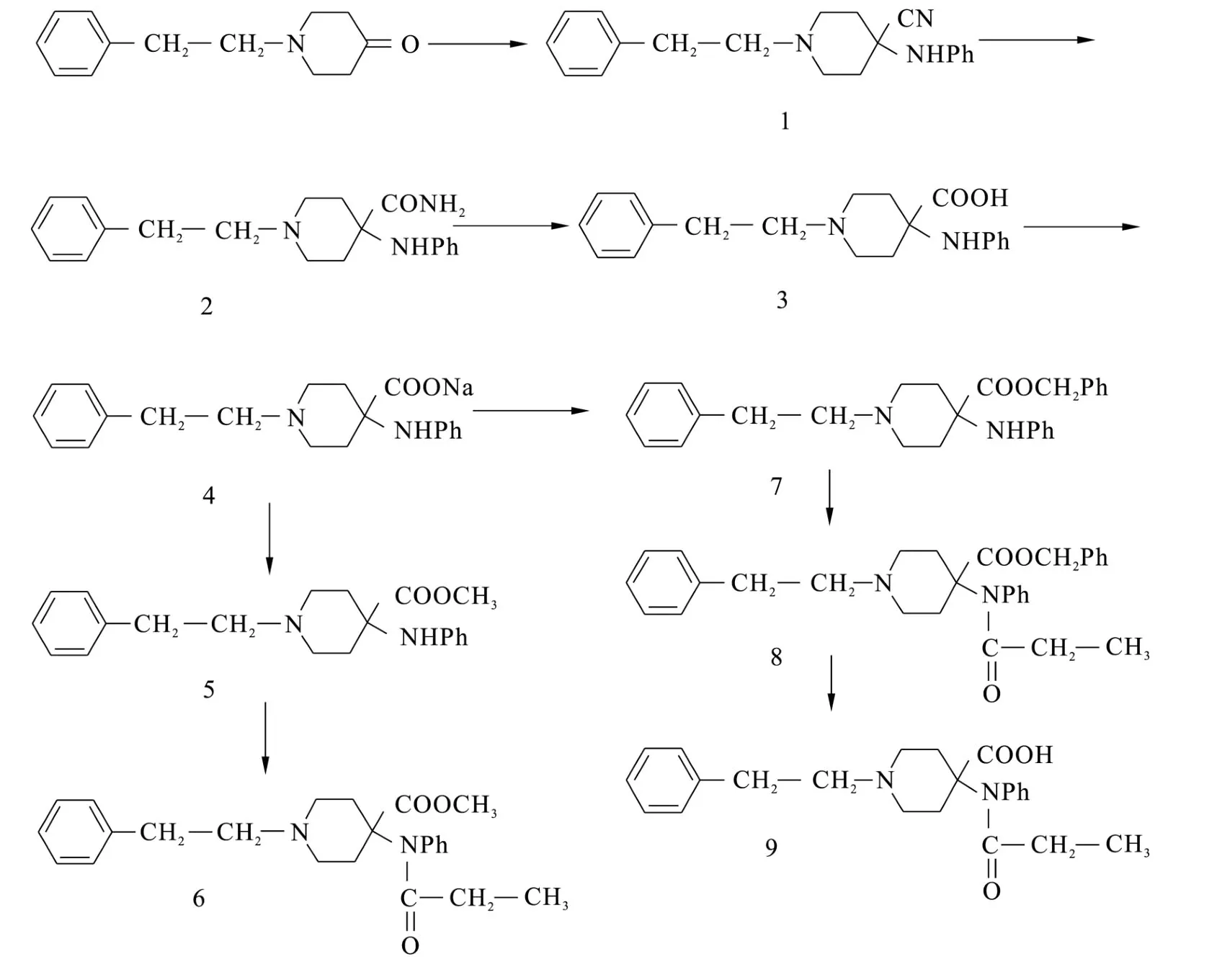
圖1 11C-CFN前體和CFN標準品合成路線Fig.1 Reaction scheme for precursor of11C-CFN and CFN reference standards
2.1.2 4-哌啶甲酰胺,4-苯胺-1-(2-苯乙基)(2)的合成 將上述5.7 g產物1慢慢加入到20 mL濃硫酸中,攪拌反應24 h后將反應混合物傾到碎冰和氨水中,氯仿萃取,提取液干燥濃縮得固體,用異丙醇洗滌得產品3.8 g。
2.1.3 4-哌啶甲酸鈉,4-苯胺-1-(2-苯乙基)(4)的合成 將2.1 g產品2、1.2 g氫氧化鉀溶于10 mL的乙二醇中,加熱回流20 h,冷卻將反應物倒入水中過濾,濾液用鹽酸酸化開始有沉淀生成,再加鹽酸后沉淀溶解,加鹽酸直到沉淀完全溶解。用濃的氫氧化鈉堿化,該反應為放熱反應,趁熱過濾除不溶物,濾液冷卻后,產物(4)結晶出來,過濾出固體再用水重結晶,得到產物0.92 g(產率41%)。
2.1.4 4-哌啶甲酸甲酯,4-苯胺-1-(2-苯乙基)(5)的合成 將0.46 g鈉鹽溶于5 mL六次甲基磷酸三酰胺(HMPA)中,加熱至70℃攪拌,然后冷卻至10℃加入碘甲烷0.22 g,反應物于室溫下繼續攪拌反應21 h。反應結束后加入5 mL甲苯,有機層用水洗滌,洗出的水相再用甲苯萃取,合并有機層,依次用w=10%的NaOH和飽和食鹽水洗滌,干燥過濾濃縮得粗產品,柱分離得產品0.17 g(產率38%),熔點95~99 ℃。
2.1.5 4-哌啶甲酸甲酯,4-[(1-丙酰基)苯胺]-1-(2-苯乙基)(6)的合成 將上述0.17 g產品5與丙酸酐1 mL混合,攪拌下回流6 h,冷卻至室溫后傾入水中,加入氨水至堿性。氯仿提取,提取液用水洗滌后干燥,過濾濃縮得油狀物,將其溶于少量異丙醇中加入草酸形成草酸鹽,過濾得白色固體0.17 g。經堿化、提取、干燥,然后減壓濃縮得到油狀物0.11 g,即為CFN標準品(產率56%)。
2.1.6 4-哌啶甲酸苯甲醇酯,4-苯胺-1-(2-苯乙基)(7)的合成 將0.46 g鈉鹽溶于5 mL六次甲基磷酸三酰胺(HMPA)中,加熱至110℃攪拌然后冷卻至20℃加入氯化芐0.15 g,反應物于室溫下繼續攪拌反應18 h。反應結束后將混合物倒入水中,用甲苯萃取,合并有機層,用飽和食鹽水洗滌,干燥過濾濃縮得粗產品,柱分離得產品0.18 g(產率32%),熔點75~78 ℃。
2.1.7 4-哌啶甲酸苯甲醇酯,1-(2-苯乙基)-4-(N-苯基丙酰胺基)(8)的合成 將上述0.18 g產品7與丙酸酐5 mL混合,攪拌下回流6 h,冷卻至室溫后傾入水中,加入氨水至堿性。氯仿提取,提取液用水洗滌后干燥,過濾濃縮得油狀物,將其溶于少量異丙醇中加入草酸形成草酸鹽,過濾得白色固體0.13 g。經堿化、提取、干燥,然后減壓濃縮得到固體0.1 g(產率45%)。
2.1.8 4-哌啶甲酸,1-(2-苯乙基)-4-(N-苯基丙酰胺基)(9)的合成 將90 mg產品8溶于10 mL無水乙醇中,加入 10%Pd/C 20 mg,(2~3)×105Pa下室溫通入氫氣脫去芐基,攪拌約16 h,過濾除去催化劑,濾餅用乙醇洗滌,將濾液濃縮得到固體物質,將其柱分離后得到最終白色固體產物14.5 mg(產率 20%)。
2.2 11C碘代甲烷的生產及11C-CH3-Triflate的合成
用 SumitomoHM-20S回旋加速器 40μA 20 MeV質子轟擊含φ=1%氧的天然氮氣靶,通過14N(p,α)11C核反應生成11C-CO2。[11C]-CH3I由單管、液相法由11C碘代甲烷儀自動化合成[9],即生成的11C-CO2由液氮捕獲后,由氮氣以30 mL/min速率載帶入0.2 mL 1 mol/L氫化鋰鋁/THF中,然后加熱除 THF,加入0.2 mLw=57%的氫碘酸,生成的11C-CH3I由氮氣載帶,進入 T riflate轉化爐;11C-CH3I經在線轉化成11C-CH3-Triflate[10]。
2.3 11C-CFN的標記及純化
1.0 mg的去甲基 CFN酸前體,用0.2 mL DMSO溶解,加入2.6μL 1 mol/L四丁基胺甲醇溶液。將上述制備的11CH3-Triflate通入該溶液。該溶液用10 mLφ=10%氨水稀釋,混合液過C-2柱,用10 mL水清洗該柱,最后用1.0 mL無水乙醇將產品從C-2柱上洗脫,加入9 mL水即可。
2.4 11C-CFN質量控制
采用 HPLC分析產品的放化純度,分析柱為C-18柱,流動相為30%MeCN/水(1L水加2 mL冰乙酸),流速為1 mL/min。11C-CFN的保留時間為5.3 min。采用LC/MS/MS測量11C-CFN的比活度[11]。首先需配制不同濃度的 CFN標準品,由LC/MS/MS測得不同濃度下CFN標準品的質量數峰值為395.2,根據濃度和峰面積作圖為線性(r2=0.999 9),依據11C-CFN在相同質量數下的峰面積,計算CFN的濃度,根據其放射化學濃度,計算11C-CFN的比活度。
3 結果和討論
3.1 CFN標準品和CFN標記前體的合成與表征
本研究合成從較易得到的原料出發,最終合成了CFN的標準品(6)和CFN的標記前體(9),從 N-(2-苯乙基)-4-哌啶酮開始計算,總體產率為0.22%。
其中化合物2:MS(m/e):324.2;1H NMR(300 MHz,DMSO,δ):7.13~7.30(m,6H),6.95~7.09(m,3H),6.52~6.63(m,3H),2.57~2.75(m,4H),2.42~2.50(M,2H),2.20~2.33(m,2H),1.90~2.04(m,2H),1.78~1.89(m,2H)。
化合物3:1H NMR(300 MHz,DMSO,δ):7.12~7.30(m,5H),6.87~6.95(m,2H),6.60~6.68(m,2H),6.32~6.41(m,1H),2.65~2.75(m,4H),2.40~2.50(m,2H),2.26~2.40(m,2H),1.92~2.05(m,2H),1.72~1.84(m,2H)。
化合物5:MS(m/e):339.2;1H NMR(300 MHz,CDCl3,δ):7.25~7.32(m,2H),7.11~7.22(m,5H),6.72~6.79(m,1H),6.55~6.62(m,2H),3.85(br,1H),3.68(s,3H),2.75~2.85(m,2H),2.57~2.75(m,4H),2.43~2.56(m,2H),2.20~2.35(m,2H),2.00~2.13(m,2H)。
化合物6:MS(m/e):395.2;1H NMR(300 MHz,CDCl3,δ):7.38~7.46(m,3H),7.30~7.35(m,2H),7.21~7.29(m,2H),7.11~7.20(m,3H),3.80(s,3H),2.67~2.80(m,4H),2.39~2.62(m,4H),2.26~2.38(m,2H),1.88(q,2H,J=7.5 Hz),1.60~1.74(m,2H),0.96(t,3H,J=7.5 Hz)。
化合物8:MS(m/e):471.2;1H NMR(300 MHz,CDCl3,δ):7.21~7.44(m,12H),7.09~7.20(m,3H),5.25(s,2H),2.63~2.75(m,4H),2.41~2.57(m,4H),2.28~2.39(m,2H),1.85(q,2H,J=7.5 Hz),1.60~1.74(m,2H),0.94(t,3H,J=7.5 Hz)。
化合物9:MS(m/e):381.2;1H NMR(300 MHz,DMSO,δ):12.30(1H),7.41~7.51(m,3H),7.35~7.40(m,2H),7.21~7.26(m,2H),7.14~7.18(m,3H),2.55~2.70(m,4H),2.38~2.50(m,4H),2.08~2.15(m,2H),1.78(q,2H,J=7.5 Hz),1.50~1.60(m,2H),0.82(t,3H,J=7.5 Hz)。
與朱國政等[7]的合成方法相比,本研究在從物質3合成5、7的時候,增加了成鹽這一步,由于成鹽后溶液的p H為中性,甲基化及芐基化時無需再加堿,甲基化時不受堿量的影響,因此產率增加,顯著提高了合成產率。
3.2 11C-CFN的標記及純化結果
在常溫、堿性條件下,前體易與11C-CH3-Triflate反應,從11C-CH3I的放射性活度計算,11C-CFN的標記率為(67.3±2.3)%(n=3)。經 HPLC分析,產品的放化純度大于98%,有二個雜質峰(圖2),一個是與溶劑相對應的 tR=2.36 min的峰,該峰為水溶性的11C-CH3-Triflate峰,另一是未知副產品的tR=4.29 min雜質峰。圖2中紫外吸收峰為共進樣CFN標準品的吸收峰(死體積已校正),該峰與放射性峰一致,說明放射性主峰tR=5.31 min即為CFN。由于采用ABX公司低本底1 mol/L氫化鋰鋁/THF,標記物的 tR=5.31 min處的紫外吸收峰很小(未列),紫外測量受干擾,經LC/MS/MS測量,11C-CFN的比活度為1.67×1014Bq/g(66.6 PBq/mol)。患者注射劑量為每人370 MBq,則單個患者接受注射的CFN量為2.2μg。CFN的安全劑量為10μg,因此,本法合成的11C-CFN放射化學純度和比活度基本滿足臨床研究的需求。
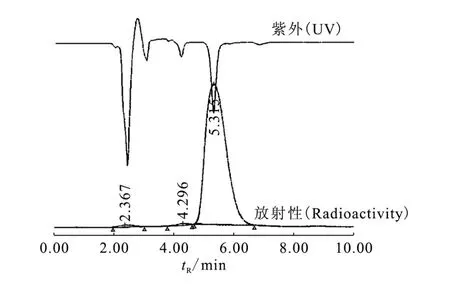
圖2 11C-CFN的HPLC分析放射性和標準品的UVFig.2 Profiles of analytical of formulation11C-CFN and UV of CFN reference standards
本方法的標記率明顯高于文獻[6]上的40%。采用11C-CH3-Triflate甲基化的優點除其活性大外,11C-CH3-Triflate本身是水溶性,而11C-CH3I為脂溶性[10],在制備中樞神經受體藥物時,可容易將產品吸附在固相柱上,未反應的11CCH3-Triflate通過水清洗固相柱達到純化的目的;而脂溶性的11C-CH3I常與產品混在一起,用固相柱不易分開。
本研究的純化方法與文獻[5-6]相比有一定區別:本方法未采用正丁醇,同時也沒有采用陰離子柱;而直接采用C-2柱吸附、清洗和洗脫,采用C-2柱可最大限度去除標記前體,少量的去甲基CFN酸前體價態為負一價,在血液中無法進入大腦血腦屏障(BBB);動物實驗證實,盡管CFN的毒性很大,但其前體去甲基CFN酸則毒性很低,注射50μg/g前體于小鼠,未觀察到小鼠中毒癥狀。采用陰離子柱的目的是提高產品的放化純度,從圖2可見,雖然沒有采用陰離子柱純化,但本產品放化純度仍達到98%。
3.3 11C-CFN的副反應
為提高11C-CFN的甲基化產率,在前體溶液中通入11CH3-Triflate后,反應液加熱至80℃反應5 min,同樣方法純化,發現產品放化純度下降,同時出現了2個脂溶性放射性峰(圖3),且tR發生了變化。經共進CFN標準品發現,tR=6.1 min的為產品,tR=4.2 min的為放化雜質,結構未知。
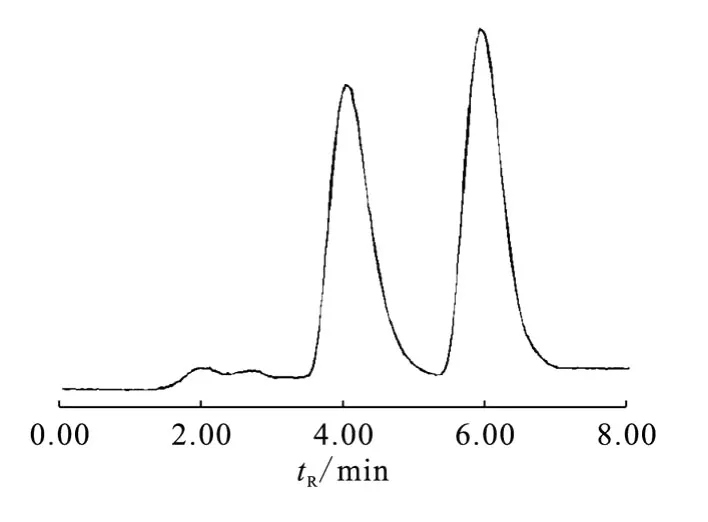
圖3 11C-CFN的副反應的HPLC峰Fig.3 Analytical HPLC of by-production of11C-CFN
在標記前體1-苯乙基-4-(N-苯基丙酰胺基)哌啶-4-羧酸上,可甲基化的位置共有3個:第一是產品的羧基位;第二是哌啶環上的氮原子,甲基化后生成季銨鹽,但本實驗采用陽離子交換柱Sep-Pak CM未能分離到該產品,可能是受溶液中其它離子干擾的緣故;第三是苯胺上的氮原子,但該原子受鄰位羰基和苯環空間位阻的影響,親核力很小。因此最有可能的副反應是第二種。本工作嘗試用質譜分析副產物,發現其分子量與產品一致,說明仍是前體甲基化產品,僅是標在了其它位置,經生物學驗證,該副產品不能進入BBB(另文報道)。
早期的文獻采用加熱的方法沒有發現這類副反應,可能是他們均采用了活性較小的碘代甲烷作甲基化試劑。因此采用高活性的11C-CH3-Triflate作甲基化試劑,無需加熱,常溫下即可發生反應;加熱后副反應會增加,需經 HPLC純化。
4 結 論
本研究從較易得的試劑1-芐基-4-哌啶酮為原料合成了11C-CFN的前體:1-苯乙基-4-(N-苯基丙酰胺基)哌啶-4-羧酸和標準品,標準品對分析副反應有很大幫助。采用11C-CH3-Triflate可在常溫下與前體反應,直接采用C-2柱吸附、清洗和洗脫,標記率為(67.3±2.3)%(n=3),產品的放化純度大于98%,比活度為1.67 ×1014Bq/g(66.6 PBq/mol)。
[1]Harris R E,Zubieta J K,Scott D J,et al.Traditional Chinese Acupuncture and Placebo (Sham)Acupuncture are Differentiated by Their Effects on Μ-Opioid Receptors(MORs)[J].Neuroimage,2009,47:1 077-1 085.
[2]Liberzon I,Taylor S F,Phan K L,et al.Altered Central Mu-Opioid Receptor Binding After Psychological Trauma[J].Biol Psychiatry,2007,61:1 030-1 038.
[3]Heinz A,Reimold M,Wrase J,et al.Correlation of Stable Elevations in Striatal Mu-Opioid Receptor Availability in Detoxified Alcoholic Patients With Alcohol Craving:A Positron Emissiontomography Study Using Carbon 11-Labeled Carfentanil[J].Arch Gen Psychiatry,2005,62:57-64.
[4]Dannals R F,Ravert H T,Frost J J,et al,Radiosynthesis of an Opiate Receptor Binding Radiotracer:[11C]Carfentanil[J].Int J Appl Radiat Isot,1985,36:303-306.
[5]Jewett D M.A Simple Synthesis of[11C]Carfentanil Using an Extraction Disk Instead of HPLC[J].Nucl Med Biol,2001,28:733-734.
[6]Shao X,Kilbourn M R.A Simple Modification of GE Tracerlab FX C Pro for Rapid Sequential Preparation of11C-Carfentanil and11C-Raclopride[J].Appl Radiat Isot,2009,67:602-605.
[7]朱國政,戴敦華.強效鎮痛藥卡芬太尼的合成[J].醫藥工業,1988,19:9-11.
[8]WadsakW. Preparationand Radiosynthesis of[18F]FE-CFN(2-[18F]Fluoroethyl 4-[N-(1-Oxopropyl)-N-Phenylamino]-1-(2-Phenylethyl)-4-Piperidinecarboxylate):A PotentialΜ-Opioid Receptor Imaging Agent[J].Radiochim Acta,2007,95:33-38.
[9]張錦明,田嘉禾,王武尚,等.單管法自動化合成11C-碘代甲烷[J].中華核醫學雜志,2004,24:243-244.
[10]張錦明,田嘉禾,王武尚,等.在線制備11C-Triflate-CH3[J].同位素,2006,19:124-128.
[11]Zhang X J,Zhang J M.Quality Control of11CCarfentanil[C]∥8thChina-Japan Seminar on Radiopharmaceutical Chemistry,Beijing:Beijing Normal University,2010:73.
Organic Synthesis of Precursor and Radiolabelling of11C-Carfentanil as Opiate Receptor Imaging Agent
ZHANGJin-ming1,GUI Yuan2,XU Zhi-hong2,ZHANG Xiao-jun1,XIANG Xiao-hui3,TIAN Jia-he1
1.Department of Nuclear Medicine,The PLA General Hospital,Beijing 100853,China;
2.Huayi Chemicals Co.Ltd.,Changshu 215522,China;
3.Neuroscience Research Institute,Peking University,Beijing 100191,China
The[11C]-methyl 1-phenethyl-4-(N-phenylpropanamido)piperidine-4-carboxylate(11C-Carfentanil)asμopiate receptor imaging agent and its precursor have been synthesized.The precursor has been confirmed by1H NMR and MS.11C-Carfentanil was prepared without using HPLC purification.Labeling yields,radiochemistry purity and specific activity are(67.3±2.3)%(n=3),98%and 1.67×1014Bq/g(66.6 PBq/mol)respectively.
11C;opiate receptor;11C-Carfentanil;PET
TL923
A
0253-9950(2011)04-0252-05
2010-07-08;
2010-12-13
國家重點研究發展計劃(973)支持項目(2007CB512500)
張錦明(1965—),男,江蘇南通人,博士,研究員,核醫學專業
猜你喜歡
現代裝飾(2022年4期)2022-08-31 01:39:32
現代裝飾(2022年3期)2022-07-05 05:55:06
物流技術與應用(2022年5期)2022-06-17 06:01:38
快樂語文(2021年36期)2022-01-18 05:48:46
金橋(2021年4期)2021-05-21 08:19:22
中國化妝品(2018年6期)2018-07-09 03:12:40
中國化妝品(2018年6期)2018-07-09 03:12:32
Coco薇(2015年1期)2015-08-13 02:23:50
汽車維修與保養(2015年6期)2015-04-17 03:31:50
玩具(2009年10期)2009-11-04 02:33:14
