氣相色譜法測定廢水中酚的研究
2011-04-12 08:14:50吳明華
科學之友 2011年8期
關鍵詞:實驗
吳明華
(東莞市大成環境檢測有限公司,廣東 東莞 523000)
隨著經濟的不斷發展,化工企業也迅速地發展。由于一些中小型企業為了減少運營成本,不顧環境保護部門的三令五申,向自然水域偷排未經處理的廢水現象屢有發生。目前,工業廢水中對揮發酚的分析方法較多,有滴定法、分光光度法、流動注射法、液相色譜法和氣相色譜法等。滴定法只能測高含量的酚,對低含量酚的測定誤差較大;分光光度法只能測定水中鄰間位取代的揮發性酚類,測定結果比實際含量偏低;流動注射法分析應用范圍窄;液相色譜法準確度高、重現性好、操作簡單,但靈敏度不高,而氣相色譜法具有靈敏度高、操作簡單、干擾小、應用廣泛等特點。這些方法多是針對揮發酚的測定,部分針對多元酚的測定。本文在已有基礎上將測定方法進行了改進,建立了可同時測定揮發酚和多元酚的方法,具有操作簡單、干擾小等優點。
1 實驗部分
1.1 儀器
GC-14C氣相色譜儀、FID檢測器、HP-5毛細柱(P/N1909lJ-4l3)30 m×320 um×0.25 um、5 mL微型注射器。
1.2 試劑
苯酚、鄰甲酚、間甲酚、對甲酚、鄰苯二酚、間苯三酚標準溶液均為1.0000 g/L,標準使用溶液均為0.1 g/L;溴溶液:稱取溴水0.1 g和溴化鉀0.16 g溶于無酚水20 mL中,配成5 g/L的溶液;抗壞血酸溶液:稱取抗壞血酸0.2 g溶于20 mL無酚水中,配成10 g/L的溶液;苯、醋酸酐、碳酸鉀、環己烷、三辛銨、磷酸三丁酯(TBP)等試劑為色譜純和分析純。
1.3 色譜條件
柱溫 200 ℃,進樣口溫度 260 ℃,氣化室溫度 300 ℃,檢測器溫度300 ℃。載氣為高純氮氣(純度為99.999%),燃氣為高純氫氣(純度為 99.999%),實驗對色譜條件進行優化選擇:柱溫從100 ℃程序升溫至200 ℃(10 ℃/min),載氣流速30 mL/min,氫氣流速30 mL/min,空氣流速400 mL/min,尾吹流速25 mL/min。
1.4 實驗方法
宜取標準溶液和工業廢水各10.0 mL,將工業廢水稀釋50倍后再分取10.0 mL,用濃度1.0 mol/L的鹽酸溶液調節樣品溶液pH至4.5左右;加溴溶液0.25 mL,振蕩3 min,再加入10 g/L抗壞血酸溶液0.25 mL,搖勻后,加入萃取劑10.0 mL,萃取1 min,靜止分層;取有機相0.8 mL注入氣相色譜儀。
2 結果與討論
2.1 色譜柱的選擇
分別選用 FFAP、DB-1、HP-1和 HP-5毛細柱(P/N19091J-413)作為酚溴化衍生物的分離色譜柱。實驗表明:FFAP毛細柱可以很好地分離4種一元酚的溴化衍生物,但對二元酚和三元酚的分離效果不理想;在DB-1、HP一1和HP-5柱中可以完全分離6種酚,但在DB-1柱一元酚和二元酚之間有干擾峰出現;在HP-1柱上分離時間較長,效果不理想。HP-5毛細柱可以較好地分離6種酚且保留時間適當,實驗中選擇HP-5毛細管柱作為色譜分離柱。
2.2 萃取劑的選擇
分別采用環己烷、磷酸三丁酯以及三辛銨和磷酸三丁酯的撮合液(按一定比例混合)作萃取劑,考查6種酚類化合物在有機相和水相中的溶解度。取10 mg/L酚混合標準溶液10 mL于50 mL萃取瓶中,按實驗方法進行操作,分離有機相和水相后,再加入萃取劑5 mL,萃取兩次,合并有機相,分別取有機相0.8 mL注入氣相色譜儀。實驗結果表明,環己烷作萃取劑時,幾種酚在環己烷中的溶解度偏低,不宜采用;磷酸三丁酯作萃取劑時,對6種酚的溶解度較好,與水的不相容性也較好,但其穩定性不理想,計算結果偏低,不宜采用;三辛銨和磷酸三丁酯(按一定比例混合)作萃取劑有較高的溶解度,能滿足微量或痕量成分的分析要求,并且分層速度很快,計算結果較準確,故本實驗選用三辛銨和磷酸三丁酯混合液作萃取劑。
同時對萃取劑的用量、鹽度進行試驗,通過在無酚水中加入已知量酚用三辛銨和磷酸三丁酯混合萃取劑萃取,選用NaC1來調節鹽度,發現當V(萃取劑)∶V(廢水)= 1∶2,鹽度為10%時,萃取效果最好。
2.3 實驗方法的選擇
實驗以苯酚、鄰甲酚、間甲酚、對甲酚、鄰苯二酚、間苯三酚的標準溶液對比了直接萃取和衍生化反應后萃取氣相色譜測定的方法。
結果表明:直接萃取法有局限,不能測定二元酚、三元酚,對一元酚的分離效果也不理想,從實驗結果看,回收率偏低。衍生化后萃取的方法可測定揮發酚和多元酚,且回收率、準確度較高,故本實驗采用衍生化后萃取再用氣相色譜測定的方法。
2.4 衍生化條件的選擇
選用醋酸酐、質量分數為10的碳酸鉀、5 g/L溴溶液對比進行衍生化試驗,同時對試劑用量、反應時間做了試驗。實驗中分別對酚標準樣和工業廢水用正交設計的方法選擇最佳衍生化反應,選擇了最佳條件進行衍生化收率的測定。試驗發現采用醋酸酐和碳酸鉀衍生化所需時間較長,用量也較大;而溴液與酚反應迅速,用時較短,用量也不大,所以實驗選用 5 g/L溴溶液進行衍生化反應,實驗選擇時間為3 min,用量為0.25 mL。
2.5 體系p H對衍生化反應的影響
一元酚和二元酚、三元酚與溴的衍生化反應速度很快,瞬間即可生成三溴酚,而三溴酚不溶于水易溶于有機溶劑,可以用有機溶劑萃取,但此反應受體系 pH的影響較大,尤其苯酚受pH影響更為顯著,見表1,表1中數值為峰面積。
可知,苯酚的溴化衍生反應pH=3~5范圍內穩定,pH大于5時體系極不穩定;鄰甲酚、對甲酚和間甲酚受體系pH的影響相對較小,在pH=3~6間峰面積出現最大值,pH大于7后其溴化衍生物的穩定性均呈下降趨勢。鄰苯二酚、間苯三酚在pH=4~7間呈現最大值。因此,綜合考慮6種酚類化合物,實驗選擇體系溴化的pH為4~5。
2.6 還原劑的選擇
在溴衍生化反應過程中,為使衍生化反應完全,必須加入過量的溴,體系中過量的溴會繼續與生成的三溴酚反應,隨著時間的增加溶液中三溴酚含量逐漸減少,影響反應結果。因此,溴化反應后,必須立即去除體系中過量的溴,故需選用還原劑將多余的溴去除。選用碘化鉀和抗壞血酸作為還原劑分別進行試驗,當體系pH為4.5,選用碘化鉀作還原劑時,生成的碘溶于有機相,使有機相呈現紅色,并且色譜圖中出現異常峰;選用抗壞血酸作還原劑時,沒有上述干擾,且體系中三溴酚在48 h內均是穩定的,故本實驗選用抗壞血酸作還原劑,向反應體系中加入10 g/L抗壞血酸溶液0~0.5 mL,可以完全消除多余的溴而不會影響反應結果。
2.7 樣品的測定及回收率實驗
按實驗方法操作,將工業廢水稀釋50倍后分取10 mL,測各組分的濃度。對同一個工業廢水樣品連續測定6次,結果見表 1,鄰甲酚、對甲酚、苯酚、間甲酚、鄰苯二酚、間苯三酚含量的相對標準偏差(n=6)分別為5.2%、4.5%、6.1%、5.9%、7.3%、7.0%。
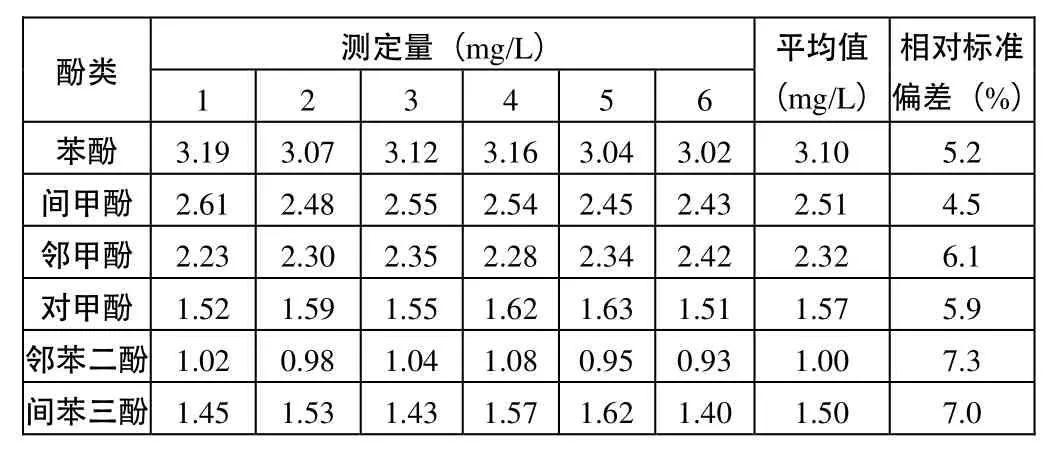
表1 工業廢水中酚類的測定結果
另取10 mL稀釋后的廢水樣分別加入10 mg/L的混合標準溶液10.0 uL和20.0 uL,注入氣相色譜儀,測定6種酚的溴化衍生物的峰面積,外標法分析計算含量,得其加標回收率為86.0%~98.1%,見表2。
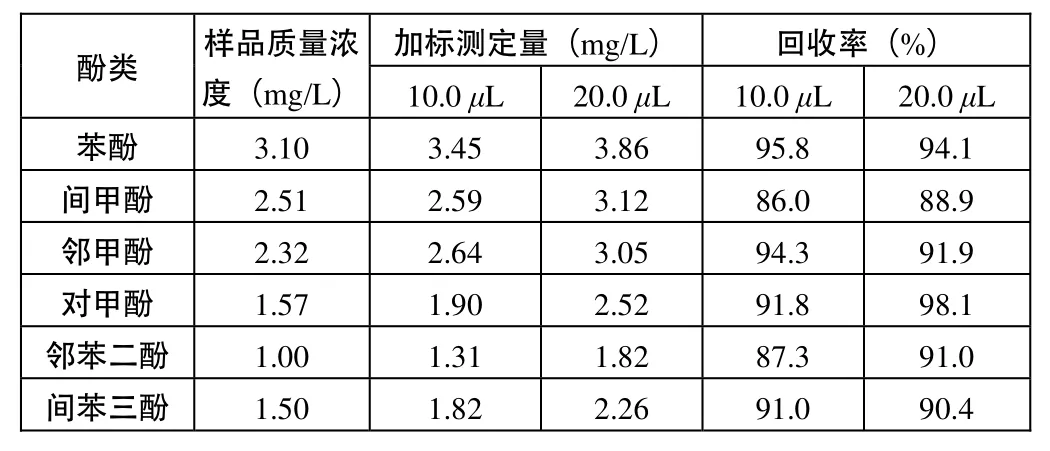
表2 工業廢水中6種酚的加標回收率試驗
3 結束語
通過實驗,充分表明該方法適用于工業廢水中揮發酚、二元酚、三元酚的定性分析及定量測定。該方法具有準確度高、重現性好、靈敏度高、干擾小的優點,而衍生化使測定范圍更廣,結果更準確,且衍生化試劑易得,操作也易控。
1 奚稼軒.固相萃取-高效液相色譜法測定飲用水中酚類化合物[J].環境化學,2004(02)
2 張明時、王愛民.溴化衍生氣相色譜法測定環境水體中痕量苯酚[J].分析化學,1999(01)
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
