新藤黃酸固體脂質納米粒的制備與質量評價
2011-07-28 05:25:38陳延杰陳衛東
中國藥業 2011年21期
陳延杰,陳衛東
(安徽中醫學院藥學院,安徽 合肥 230038)
固體脂質納米粒(solid lipid nanoparticles)給藥系統是目前亞微粒給藥系統的發展熱點。它以生理相容的高熔點脂質為骨架材料制備,將藥物包裹或內嵌于類脂核中,制成粒徑約為50~1000 nm的固體膠粒給藥系統,副作用小,室溫及體溫下呈固態粒子[1-2]。新藤黃酸(neo-gambogid acid)是從中藥藤黃中分離出的一種具有較高抗腫瘤活性的化合物,抗癌譜廣,對人肝癌細胞Bel-7402、人非小細胞肺癌細胞A549、人結腸癌細胞HCT-8以及人卵巢癌細胞A2780的增殖均有明顯抑制作用[3]。但新藤黃酸水溶性差,而且靜脈給藥時有強烈的血管刺激性,限制了其臨床應用。此外,新藤黃酸在體內的半衰期短,對治療效果也有很大的影響。結合臨床需求和目前制劑的發展情況,筆者將新藤黃酸藥制備成固體脂質納米粒,控制粒徑大小,期望減小新藤黃酸的血管刺激性,提高體內生物利用度,更好地發揮其藥理作用。
1 儀器與試藥
島津高效液相色譜儀(LC-20AB泵,SPD-M20A紫外檢測器及島津LC-solution色譜工作站);H-800型透射電子顯微鏡(日本日立公司);Delsa nano C型粒度分析儀(美國Beckman Coulter公司);UV-2550型紫外可見分光光度計(日本島津公司);CP225D型電子天平(德國賽多利斯公司);85-2型恒溫磁力攪拌器(江蘇金壇市環宇科學儀器廠);HH-S2型數顯恒溫水浴鍋(江蘇金壇市環宇科學儀器廠)。新藤黃酸原料藥(安徽中醫學院藥化教研室提供);卵磷脂(安徽豐原藥業股份有限公司提供);泊洛沙姆188(德國BASF公司,批號為WO39846);單硬脂酸甘油酯(汕頭市西隴化工廠有限公司,批號為0906012);吐溫-80(國藥集團化學試劑有限公司,批號為F20050721);葡聚糖凝膠G-50(美國Pharmacia公司);其他試劑均為分析純。
2 方法與結果
2.1 新藤黃酸固體脂質納米粒制備
取處方量的新藤黃酸、單硬脂酸甘油酯、卵磷脂溶于適量無水乙醇中,于(75±2)℃水浴下形成有機相。另取泊洛沙姆溶于蒸餾水中,加入少量吐溫-80,加熱至與有機相相同溫度,構成水相。在攪拌過程中用針頭將有機相注入水相,按照每秒1滴的速度,整個過程中反應溫度要保持在脂質材料的熔點之上。攪拌1 h,待無水乙醇揮干,將所得的半透明體系快速分散于0~2℃的冰水中,并且冰浴攪拌30 min,即得納米粒混懸液。
2.2 外觀與粒徑
按照優化后的處方制備樣品,將樣品用質量分數為2%的磷鎢酸溶液負染,然后滴至銅網上,室溫放置至形成薄膜后,用透射電子顯微鏡觀察。由圖1可見,新藤黃酸固體脂質納米粒為具有黃色乳光膠體溶液,電鏡下觀察納米粒為許多圓形或橢圓形球粒,平均粒徑為(113±6)nm。
2.3 包封率測定
2.3.1 檢測波長選擇
精密稱取新藤黃酸2 mg對照品,用甲醇溶解稀釋,定容到10 mL容量瓶中,振搖作為新藤黃酸濃貯備液。以濃貯備液濃度配制質量濃度為0.2 g/mL的新藤黃酸標準溶液,同法配制空白溶液作參比,用紫外分光光度計于200~600 nm波長范圍內掃描,結果新藤黃酸在360 nm波長處有最大吸收,故選擇360 nm為檢測波長。
2.3.2 色譜條件
色譜柱:Lichrospher C18柱(250 mm ×4.6 mm,5 μm);流動相:甲醇-0.1% 磷酸(9 ∶1);流速:1.0 mL/min;柱溫:25 ℃;檢測波長:360 nm;進樣量:20 μL。
2.3.3 方法學考察
專屬性試驗:取新藤黃酸對照品溶液、空白固體脂質納米粒破乳溶液和新藤黃酸固體脂質納米粒破乳溶液各20 μL進樣分析,色譜圖見圖2。結果顯示輔料對藥物含量測定無干擾。
線性關系考察:精密吸取不同體積的貯備液,流動相分別稀釋成 50,25,10,1.0,0.1μg/mL 的標準溶液,分別取 20 μL 進樣,在按2.2.2項下色譜條件進行測定,計算峰面積。結果表明,以峰面積(A)為縱坐標、質量濃度(C,μg/mL)為橫坐標進行線性回歸分析,得標準曲線方程 A=30992 C-18289,r2=0.9995。
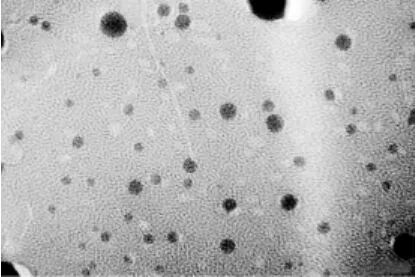
圖1 透射電鏡圖
圖2 高效液相色譜圖
加樣回收試驗:按處方比例加入1 mL空白固體脂質納米粒溶液至編號為 1,2,3的10 mL量瓶中,依次精確加入上述新藤黃酸貯備液 0.5,1,2 mL,以甲醇稀釋至刻度,得質量濃度10~40μg/mL的樣品溶液,分別進樣20μL,測定峰面積,連續3次,代入標準曲線換算成實際測定濃度,計算回收率。結果平均加樣回收率為99.3%,RSD為1.12%。
精密度試驗:取同一新藤黃酸對照品溶液,連續進樣5次,測定峰面積。結果峰面積的 RSD為1.02%,表明方法精密度良好。
包封率測定[4]:以Sephadex G-50葡聚糖凝膠柱色譜法分離和游離藥物,測定其包封率。吸取1 mL新藤黃酸固體脂質納米粒混懸液上柱,蒸餾水洗脫,收集帶有乳光部分的洗脫液,每2 mL收集1次,加甲醇并超聲破乳。另吸取1 mL新藤黃酸固體脂質納米粒混懸液,直接加甲醇破乳并超聲提取。按2.3.2項下色譜條件分別測定兩樣品中新藤黃酸的含量,按公式包封率=m柱/m總計算包封率。m柱為新藤黃酸固體脂質納米粒混懸液通過Sephadex G-50后測得的新藤黃酸的含量;m總為新藤黃酸固體脂質納米粒混懸液直接破乳后測得的新藤黃酸的含量。按處方量進行了3次重復驗證試驗,制備 3批固體脂質納米粒,得平均包封率為(60.1±1.1)%。
2.4 正交試驗與方差分析
在單因素考察試驗結果的基礎上,確定單硬脂酸甘油酯(因素A)、泊洛沙姆用量(因素B)、乳化時間(因素C)、單硬脂酸甘油酯與卵磷脂比(因素D)4個因素,每個因素設定3個水平,進行正交設計試驗,以包封率為考察指標篩選最佳處方及工藝參數。因素水平設計見表1,試驗結果見表2和表3。可知,本試驗4個因素影響程度為A>D>C>B,方差分析結果表明,A和B因素有顯著影響,初步確定最佳制備工藝為A2B1C1D3,即單硬脂酸甘油酯15 mg,泊洛沙姆濃度為0.8%,攪拌時間1 h,卵磷脂30 mg。

表1 因素水平設計表
2.5 工藝的驗證與確定
根據2.4項下確定的最優處方和工藝條件平行制備3批載藥固體脂質納米粒,分別測定其包封率和載藥量,確定乳化蒸發-低溫固化法的工藝流程,結果見表4。驗證結果表明最佳處方重復性強,包封率和載藥量保持穩定。
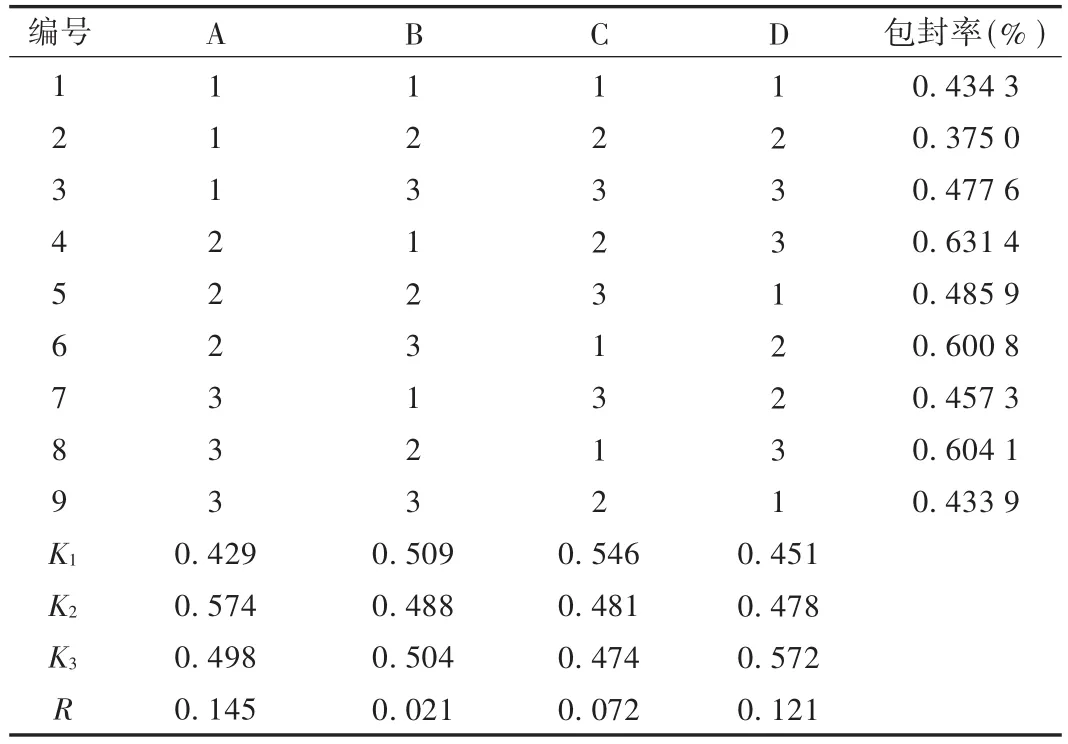
表2 正交試驗結果
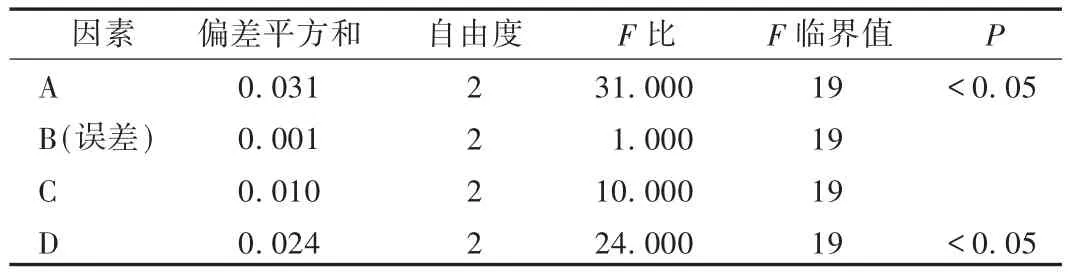
表3 方差分析結果
2.6 納米粒粒徑與電位的測定
取新藤黃酸固體脂質納米粒混懸液適當稀釋后,用納米激光粒度儀測定粒徑及分布,結果見圖3、圖4。結果表明制得的平均粒徑為163.3 nm,粒徑大小相近,Zeta電位為-16.9 mV。

表4 重復最佳處方制備的固體脂質納米粒的包封率和載藥量(%)
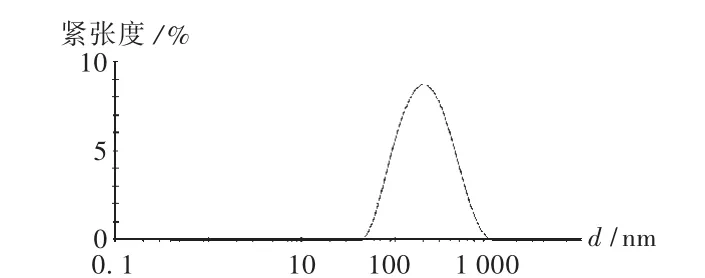
圖3 新藤黃酸固體脂質納米粒粒度分布圖
3 討論
本試驗采用高溫乳化-低溫固化法制備新藤黃酸固體脂質納米粒,載體材料為單硬脂酸甘油酯和卵磷脂,表面活性劑以泊洛沙姆為主,吐溫-80為輔,兩者混合使用可得到比較理想的固體脂質納米粒,控制粒徑在100~200 nm范圍內。

圖4 新藤黃酸固體脂質納米粒Zeta電位圖
單硬脂酸甘油酯的熔點在55℃左右,在制備過程中反應溫度應高于熔點,最終確定反應溫度為(75±2)℃ 。制備初乳時,攪拌速率對反應過程有較大的影響:攪拌速率過低,反應不完全,有機溶劑揮發不完全,造成有溶劑殘留;而轉速過大,乳化時會產生大量泡沫,影響表面活性劑的效果,最終確定轉速為1000 r/min。該方法固化的過程實際也是稀釋的過程,故所得混懸液的固體含量較低,粒子比較分散。
試驗中發現,固體脂質納米粒穩定性不理想,放置一段時間后會有沉淀析出,可能是受周圍光線、溫度、濕度影響,造成晶型的改變[5],從β'轉變成β1,使晶格轉變,導致部分沉淀在混懸液中析出。期待進一步試驗,將固體脂質納米粒混懸液做成凍干粉,以加強它的穩定性。
[1]Schwarz C,Mehner W,Lucks JS,et al.Solid lipid nanopartieles(SLN)for controlled drug delivery.Production,characterization and sterilization[J].Control Release,1994,30:83-96.
[2]Zur Muhlen A,Schwarz C,Mehnert W.Solid lipid nanoparticles(SLN)for controlled drug delivery-drug release and release mechanism[J].Eur J Pharm Biopharm,1994,45:149-155.
[3]程 卉,彭代銀,王效山,等.新藤黃酸體內外抗腫瘤作用研究[J].中草藥,2008,3(2):236-240.
[4]張 洪,詹新安,成 蓓,等.聯苯雙酯固體脂質納米粒的制備[J].廣東藥學院學報,2007,23(6):37-40.
[5]Freitas C,Muller RH.Stability determination of solid lipid nanoparticles(SLN)in aqueous dispersion after addition of electrolyte[J].J Microencapsul,1999,16(1):59-71.
