利用分子標記早期篩選光皮樺核心種質
2011-07-30 10:58:04李海英梁志偉黃華宏童再康盧泳全
浙江林業科技 2011年3期
關鍵詞:資源
李海英,梁志偉,陳 沖,黃華宏,童再康,盧泳全
(浙江農林大學 亞熱帶森林培育國家重點實驗室培育基地,浙江 臨安 311300)
植物遺傳資源是物種多樣性保護及品種選育的重要物質基礎。理論上保存的遺傳資源越多越好,但種質資源保存過多會耗費巨大的人力物力,也難以對所保存的材料進行合理評價,制約了種質的利用。因此,Frankel于1984年提出了核心種質(core collection)的概念[1],即以最少數量的種質材料代表一個物種及其近緣野生種最大限度的遺傳多樣性。核心庫中入選的遺傳材料都要有代表性,并包括盡可能多的遺傳多樣性。
構建核心種質庫主要涉及兩方面的內容,一是構建核心種質所采用的抽樣方法;二是核心種質遺傳多樣性的評價指標。在傳統的方法中,一般是以形態學及農藝性狀為評價指標[2~4]。但是,由于木本植物形態學性狀觀察周期長,與經濟價值相關的材性性狀往往需要幾十年的時間才能準確觀測,因而制約了多年生木本植物核心種質庫的構建。目前,只在白樺[5]、臘梅[6]和棗[7]等幾個物種中有相關報道。
近年來,隨著生物技術的迅猛發展,分子標記越來越多地應用到種質資源研究中。由于分子標記是以個體間遺傳物質—核苷酸序列變異為基礎的遺傳標記,是DNA水平遺傳變異的直接反映。因此,在生物發育的不同階段,不同組織的DNA都可用于遺傳資源的標記分析[8]。由于分子標記所揭示的多態性不受外界環境和內在發育階段的影響,所以非常適于多年生木本植物種質資源的早期/幼苗期鑒定。劉勇等[9]利用分子標記技術確立了柚的核心種質資源。研究結果表明,核心種質能代表初始種質群。
光皮樺(Betula luminifera)是我國南方山地常見的優良速生用材闊葉樹種,耐貧瘠,生長快,材質好,是一種極具推廣價值的造林樹種。本研究于2005年和2006年分別從浙江、廣西、福建、貴州四省的9個光皮樺天然種源采集種子,溫室內種植。實生苗生長1 a后,采優株穗條嫁接于浙江農林大學光皮樺種質園。目前,該種子園共有材料519份。本研究旨在利用分子標記技術,從這些資源中篩選具有代表性的核心資源,為光皮樺種質資源的合理保存及早期利用奠定理論基礎。
1 材料與方法
1.1 初始種質的選取
本實驗采用分組取樣法,根據種源差異將整個群體分為9個組(見表1)。然后組內采用隨機取樣,結合組內個體的表現型差異大小確定各組的取樣比例。最終,從519份共9組光皮樺樣本中抽取62份作為初始種質。于2009年5月,取正常生長的嫩葉,采用CTAB+硅珠吸附法[10]提取葉片總DNA。
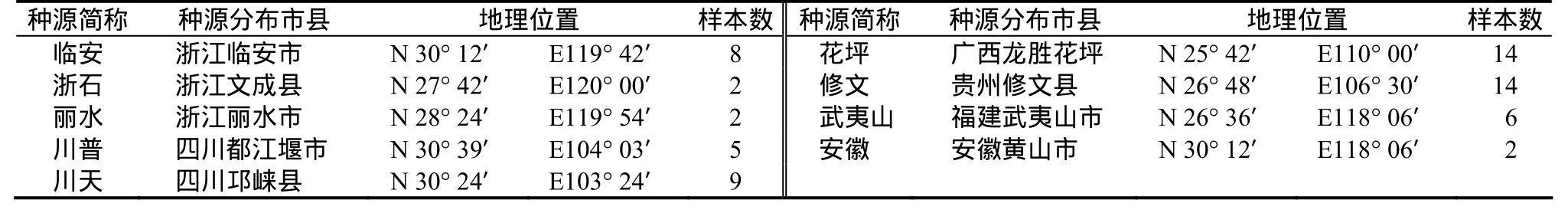
表1 光皮樺種源地理位置Table 1 Geographic location of samples
1.2 分子標記方法
采用由本實驗室開發的擴增共有序列遺傳標記(Amplified Consensus Genetic Markers, ACGM)分子標記[11]對光皮樺進行遺傳多樣性分析,共得到137個多態性位點。以1和0記錄等位基因的有和無,獲得矩陣。利用系統分析軟件POPGENE 32計算居群的多態位點數、多態位點百分率(percentage of polymorphic belt, PPB%)、觀測等位基因數(Na)、有效等位基因數(Ne)、Nei基因多樣性指數和 Shannon信息指數[9],按類平均數聚類方法(UPGMA)進行聚類,得到UPGMA系統樹。
1.3 柚類核心種質構建方法
利用ACGM所得到的譜帶進行聚類分析,按照聚類分析結果并結合形態學性狀,將遺傳相似系數最大的2個種質刪除1份,剩余的種質再聚類;再從遺傳相似系數最大的成對種質中刪除一份。以此類推,直到代表性或核心種質量達到要求。利用上述方法分別抽取41、36、27、18和9個樣品組成核心樣本群進行分析比較,各樣品群代號分別為I、II、III、IV、V。
樣品群 I(41 個樣本)代號為:2,3,4,5,6,7,9,10,11,12,13,14,15,16,17,18,20,21,22,26,27,28,30,31,37,38,41,43,44,46,47,48,50,57,58,59,60,61,62,64,65。
樣品群 II(36 個樣本)代號為:3,4,5,6,7,9,10,11,12,13,14,15,16,17,18,20,21,26,27,30,31,37,38,41,43,44,46,47,48,58,59,60,61,62,64,65。
樣品群 III(27 個樣本)代號為:4,5,6,9,10,11,14,15,16,17,18,21,27,30,31,41,43,44,48,58,59,62,64,65。
樣品群IV(18個樣本)代號為:4,5,9,10,11,14,15,17,18,27,30,31,43,44,58,59,64,65。
樣品群V(9個樣本)代號為:5,10,14,17,27,30,44,58,64。
1.4 核心種質的評價
構建好核心種質后,用多態位點數、多態位點百分率、觀測等位基因數、有效等位基因數、Nei基因多樣性指數和Shannon信息指數評價指標來評價核心種質。利用Microsoft Excel中的分析數據庫,對初始種質和核心種質的觀測等位基因數、有效等位基因數、Nei基因多樣性指數和Shannon信息指數進行t檢驗[9],根據檢驗結果來評價核心種質。
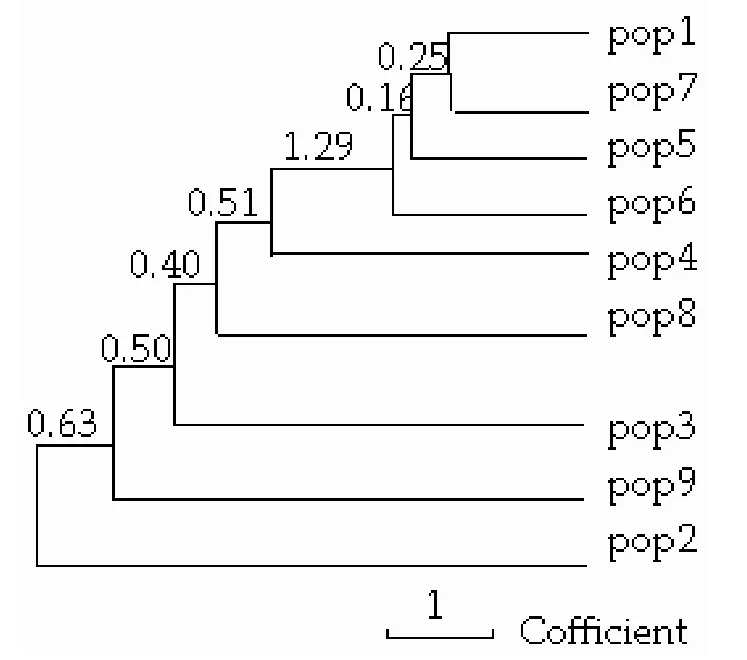
圖1 9個居群UPGMA聚類Figure 1 UPGMA dendrogram among 9 populations
2 結果與分析
2.1 初級種質的遺傳多樣性分析
通過PopGen32[9]軟件分析得到居群聚類結果(圖1),臨安(pop1)和修文(pop7)最先聚在一起,然后川天(pop5)、花坪(pop6)、川普(pop4)、武夷山(pop8)、麗水(pop3)、安徽(pop9)和浙石(pop2)依次相聚。從光皮樺種源的地理分布上看,修文(pop7)、川天(pop5)、花坪(pop6)和川普(pop4)位于我國西南地區的貴州、四川和廣西三省,而武夷山(pop8)、麗水(pop3)、安徽(pop9)和浙石(pop2)來源于我國東南地區的福建、浙江和安徽三省。由此可見,本研究的聚類結果與光皮樺各種源的地理分布是相符的。
2.2 各樣本群的遺傳多樣性比較
利用 PopGen32軟件分析各樣本群的多態性位點數、多態性位點百分率、觀測等位基因數、有效等位基因數、Nei遺傳多樣性指數和Shannon信息指數(結果見表2)。比較不同取樣數目所獲得的各種遺傳參數,結果表明,樣品群II的有效等位基因數是最高的,多態性位點數、多態性位點百分率、觀測等位基因數、Nei遺傳多樣性指數和Shannon信息指數等僅比樣品群I的稍低,考慮到取樣數的問題,我們把樣本群II列為核心種質。最后獲得的核心種質的編號為:3,4,5,6,7,9,10,11,12,13,14,15,16,17,18,20,21,26,27,30,31,37,38,41,43,44,46,47,48,58,59,60,61,62,64,65。

表2 各個樣品群的遺傳多樣性分析Table 2 Analysis of genetic diversities among different sampling groups
2.3 核心種質的評價
用 PopGen32軟件比較初始種質與核心種質的觀測等位基因數、有效等位基因數、Nei遺傳多樣性指數、Shannon 信息指數和多態性位點百分率(結果見表3),并對兩個群體的各參數進行t測驗(結果見表4)。從表3可看出核心種質保留了初始種質58.06%的樣品,多態性位點保留率達到了96%,觀測等位基因數、有效等位基因數、Nei遺傳多樣性指數和Shannon信息指數的保留率分別為 98.58%、99.88%、98.74%、98.30%。由 t測驗的結果可表明,核心種質的觀測等位基因數、有效等位基因數、Nei遺傳多樣性指數和Shannon信息指數在概率0.01水平上與初始種質差異不顯著,核心種質能很好的代替初始種質。
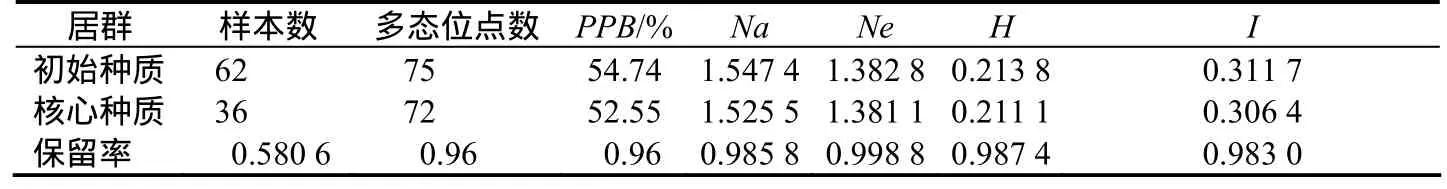
表3 初始種質與核心種質遺傳多樣性對比Table 3 Comparision of the genetic diversities between primary sample and core collection
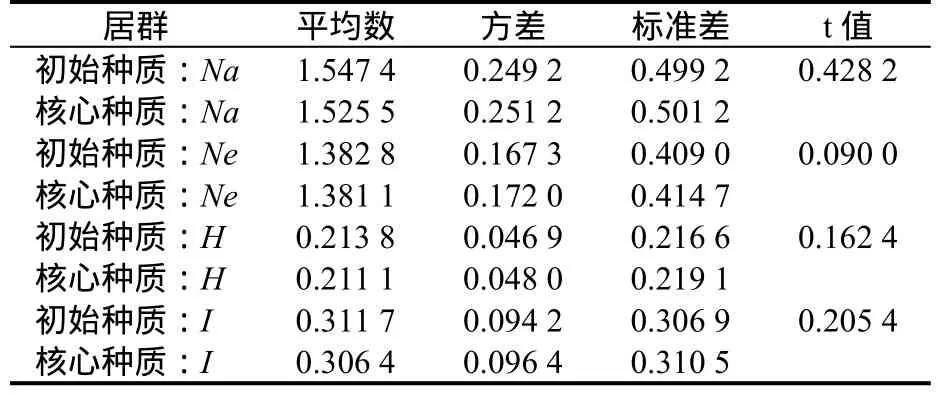
表4 初始種質與核心種質t測驗結果Table 4 t-test result between initial collection and core collection
3 討論
在構建核心種質時,抽樣比例很重要。目前,多數植物核心種質的抽樣比例為全部收集種質的5% ~ 30%,一般為10%左右[12]。但是因為生物進化及人工選擇對作物干預的存在,產生了各個物種的特性,所以對整體的取樣比例不能簡單格式化,應視研究作物的遺傳結構及遺傳多樣性而定。在本研究中,構建初始種質時,抽樣比例為12%,與多數植物核心種質的抽樣比例適當。但是在構建核心種質時,因為光皮樺初始種質的樣本數量較少,只有62份,所以得到的核心種質的比例為初始種質的58.06%。
抽樣技術也很重要,直接關系到核心庫的好壞。常用的組內取樣法有按分層抽樣[13]、遺傳差異取樣[14]、組內隨機取樣[15]、多次聚類抽樣[16]等方法。本實驗中采用組內隨機取樣,先按聚類分析分組,再從各組隨機抽取樣品得到光皮樺核心種質,最后通過 t檢驗,證明本研究得到的核心種質能很好的代表原光皮樺資源群體的遺傳變異和遺傳結構。
[1]Frankel O H. Genetic Manipulation : Impact on Man And Society[M]. Cambridge Uni Press,1984. 161-170.
[2]Brown A H D. The use of plant genetic resources [A]. Brown A H D,Frandel O H,Marshall R D,et al. [C]. England: Cambridge Univ Press,1989.
[3]Corley Holbrook C, William F Anderson. Evaluation of a core collection to identify resistance to late leafspot in peanut[J]. Core Science,1995(35):1700-1702.
[4]沈金雄,郭慶元,張秀榮,等. 中國芝麻種質資源的聚類分析[J]. 華中農業大學學報,1995,14(6):532-536.
[5]魏志剛,高玉池,劉桂豐,等. 白樺核心種質的初步構建[J]. 林業科學,2009,45(10):74-78.
[6]趙冰. 臘梅種質資源遺傳多樣性與核心種質構建的研究[D]. 北京:北京林業大學,2008.
[7]董玉慧. 棗樹農藝性狀遺傳多樣性評價與核心種質構建[D]. 石家莊:河北農業大學,2008.
[8]李麗,何偉明,馬連平,等. 用EST-SSR分子標記構建大白菜核心種質及其指紋圖譜[J]. 基因組學與應用生物學,2009,28(1):76-88.
[9]劉勇,孫中海,劉德春,等. 利用分子標記技術選擇柚類核心種質資源[J]. 果樹學報,2006,23(3):339-345.
[10]張博,張露,諸葛強,等. 一種高效的樹木總DNA的提取方法[J]. 南京林業大學學報(自然科學版),2004,28(1):13-15.
[11]Yongquan Lu,Haiying Li,Xi Chen,et al. Development amplication concensus genome markers in Betula luminifera based on Birch EST database[J]. Wood Res, 2011, 56(2):169-178.
[12]李自超,張洪亮,曹永生,等. 中國地方稻種資源初級核心種質取樣策略研究[J]. 作物學報,2003,29(1):20-24.
[13]劉三才,鄭殿升,曹永生,等. 普通小麥核心種質抽樣方法的比較[J]. 麥類作物學報,2001,21(2):42-45.
[14]徐海明,胡晉,朱軍. 構建作物種質資源核心庫的一種有效抽樣方法[J]. 作物學報,2000,26(2):157-162.
[15]魏興華,顏啟傳,應存山,等. 建立浙江地方秈稻種資源的核心樣品研究[J]. 中國水稻學,1999,13(2):81-85.
[16]HU J,Zhu J,Xu H M. Methods of constructing core coolletions by stepwise clustering with three sampling strategies based on the genotypic values of crops[J]. Theor Appl Genet,2000(101):264-268.
猜你喜歡
江蘇安全生產(2023年1期)2023-02-08 05:58:38
資源節約與環保(2022年8期)2022-09-20 02:25:22
吉林廣播電視大學學報(2021年4期)2022-01-14 02:35:48
藝術品鑒(2020年7期)2020-09-11 08:04:44
作文成功之路·小學版(2020年5期)2020-06-11 12:48:26
東方少年·布老虎畫刊(2020年4期)2020-06-08 15:48:10
小天使·一年級語數英綜合(2018年11期)2018-11-23 09:47:26
當代貴州(2018年28期)2018-09-19 06:39:04
資源再生(2017年3期)2017-06-01 12:20:59
決策(2015年9期)2015-09-10 07:22:44
