甘肅當歸炮制前后HPLC特征圖譜及阿魏酸含量測定研究
2011-08-07 05:58:58高昌琨嚴安定安徽醫科大學第一附屬醫院藥劑科合肥市230022
中國藥房 2011年47期
張 永,高昌琨,嚴安定(安徽醫科大學第一附屬醫院藥劑科,合肥市 230022)
當歸作為常用中藥材,始載于《神農本草經》,被列為中品,已有2 000多年的藥用歷史。當歸為傘形科植物當歸Angelica sinensis(Oliv.)Diels的干燥根,具有補血活血、調經止痛、潤腸通便之功效[1],主產于甘肅、云南、陜西、四川等地。甘肅是當歸的傳統道地產區,產量約占全國總產量的90%。甘肅當歸以產量高、質量優而享有盛譽,特別是“岷歸”更是馳名古今、蜚聲海外。由于當歸的質量除了受土壤、光照、氣候等環境因素影響外,還與藥材的品種、產地、采收加工等因素密切相關,因此不同產地的當歸質量各不相同[2];即使同一產地的當歸,其質量也有一定的差異[3]。而且,當歸在臨床上常以炮制品入藥,不同的炮制過程對其質量的影響也各不相同[4]。因此,有必要建立一種評價當歸炮制過程質量變化的方法,以全面評價當歸不同炮制品的質量,保證飲片療效。
本研究中,筆者以甘肅3個主產地的當歸為研究對象,利用特征圖譜技術[5]建立了甘肅當歸的高效液相色譜(HPLC)特征圖譜分析方法,并在相同色譜條件下對炮制前后甘肅當歸中阿魏酸的含量進行了測定,建立了更加全面合理的中藥飲片鑒別和質量評價方法。
1 儀器與試藥
LC-20AB HPLC儀,包括DGU-20A3在線脫氣機、SIL-20A自動進樣器、SPD-M 20A檢測器、CTO-20A柱溫箱和Class-VP色譜工作站(日本島津公司);KQ5200DB數控超聲波清洗器(功率:250W,頻率:40 kHz,昆山市超聲儀器有限公司);R-210旋轉蒸發儀(瑞士BUCHI公司);AL104電子天平(梅特勒-托利多儀器(上海)有限公司);高速萬能粉碎機(天津市泰斯特儀器有限公司)。
甲醇、乙腈為色譜純(美國Merck公司),其他試劑均為分析純;阿魏酸標準品(南京澤朗醫藥科技有限公司,純度>98%);收集3個主產地當歸藥材各50 kg,分別產自甘肅漳縣(批號:091128)、甘肅渭源(批號:090606)、甘肅岷縣(批號:090605),均由合肥和義堂中藥飲片有限公司提供,經安徽中醫學院劉守金教授鑒定為傘形科植物當歸A.sinensis(Oliv)Diels的干燥根,標本保存于安徽醫科大學第一附屬醫院藥劑科,標 本編號 分別為 AMR-091128、AMR-090606、AMR-090605。
2 方法與結果
2.1 當歸片與酒當歸的炮制加工
不同產地的甘肅當歸樣品均由合肥和義堂中藥飲片有限公司在生產條件下加工炮制成當歸片和酒當歸。取不同產地的當歸藥材,清洗后用水淋洗軟化,用切藥機切成1.5mm的薄片,置烘箱中50℃干燥4 h至干,即得當歸片;取當歸片,按100 kg當歸片加10 kg黃酒的比例加黃酒拌勻、潤透,然后置炒藥機中95℃炒制20m in至干,顏色呈深黃棕色,即得酒當歸。具體炮制加工流程見圖1。
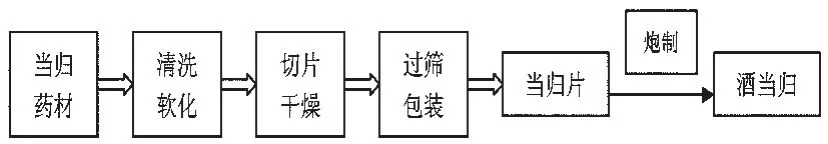
圖1 當歸炮制加工過程示意圖Fig 1 Flow chartof A.sinensis processing
2.2 色譜條件
色譜柱:Agilent HC-C18(250mm×4.6mm,5μm);柱溫:35 ℃;流速:1.0m L·min-1;檢測波長:254 nm;流動相:乙腈(A)-0.2%乙酸水溶液(B),梯度洗脫(0~30min,5%~18%A;30~65m in,18%~38%A;65~75min,38%~50%A;75~85 min,50% ~56%A;85~95 min,56%A;95~110 min,56%~65%A;110~120 min,65%~85%A;120~125 min,85%~5%A);進樣量:10μL。
2.3 標準品溶液的制備
精密稱取阿魏酸標準品5.0mg,置于5m L量瓶中,加甲醇溶解并稀釋至刻度,得濃度為1.000mg·m L-1的阿魏酸標準品溶液,冷藏備用。
2.4 供試品溶液的制備
精密稱取當歸樣品粉末1.0 g,置100m L具塞錐形瓶中,加甲醇30m L,密塞,超聲提取30m in,取出放冷,用濾紙濾過,藥渣連同濾紙放回錐形瓶中,再加甲醇30m L,同法再超聲提取1次,放冷,用濾紙濾過,合并2次濾液,減壓蒸干,殘渣用甲醇溶解并轉移至10m L容量瓶中,加甲醇稀釋至刻度,搖勻,即得。進樣前以0.45μm微孔濾膜過濾。
2.5 甘肅當歸炮制前后特征圖譜的建立
2.5.1 精密度試驗 取甘肅渭源當歸供試品溶液適量,按“2.2”項下色譜條件連續進樣6次,以阿魏酸峰為標準峰,計算得各共有峰相對保留時間的RSD均<0.118%,相對峰面積的RSD均<2.13%,表明方法精密度良好。
2.5.2 穩定性試驗 取甘肅渭源當歸供試品溶液適量,分別于0、4、8、12、16、24 h進樣分析。結果,各共有峰相對保留時間的RSD均<0.109%,相對峰面積的RSD均<2.14%,表明供試品溶液在24 h內穩定性良好。
2.5.3 重復性試驗 取甘肅渭源當歸樣品適量,按“2.4”項下方法平行制備6份供試品溶液,照“2.2”項下色譜條件分別進樣分析。結果,各共有峰相對保留時間的RSD均<0.138%,相對峰面積的RSD均<5%,表明方法重復性良好。
2.5.4 特征峰的指認 精密吸取阿魏酸標準品溶液5μL,注入色譜儀,按“2.2”項下色譜條件進樣分析,確定在此色譜條件下阿魏酸的位置(圖2);同時,以甲醇為空白溶劑進樣5μL,確認空白溶劑中相應位置沒有干擾峰存在。
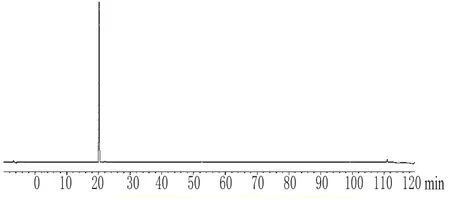
圖2 阿魏酸的HPLC圖Fig 2 HPLC chromatogramsof ferulic acid
2.5.5 甘肅當歸炮制前后特征圖譜的測定 分別精密稱取甘肅漳縣當歸片與酒當歸、甘肅渭源當歸片與酒當歸、甘肅岷縣當歸片與酒當歸樣品適量,分別按“2.4”項下方法制備供試品溶液,照“2.2”項下色譜條件進樣分析,建立各樣品的特征圖譜,詳見圖3。
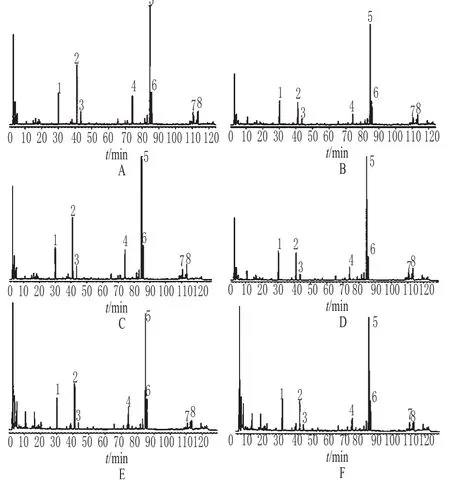
圖3 甘肅當歸炮制前后HPLC特征圖譜A.漳縣當歸片;B.漳縣酒當歸;C.渭源當歸片;D.渭源酒當歸;E.岷縣當歸片;F.岷縣酒當歸;1~8.共有峰;1.阿魏酸Fig 3 HPLC specific chromatogram s of crude and processed A.sinensis A.crude A.sinensis from Zhangxian;B.processed A.sinensis from Zhangxian;C.crude A.sinensis from Weiyuan;D.processed A.sinensis from Weiyuan;E.crude A.sinensis from M inxian;F.processed A.sinensis from M inxian;1~8.common peaks;1.ferulic acid
由圖3可以看出,甘肅不同產地當歸的HPLC特征圖譜中各成分的種類差別不大,采用特征圖譜技術可以有效鑒別甘肅當歸藥材。但甘肅不同產地當歸炮制前后的HPLC特征圖譜中某些成分的含量發生了明顯變化,如炮制后2、3、4號峰含量明顯降低,1、6號峰含量略有降低,說明炮制對當歸成分含量具有一定的影響。
2.6 甘肅當歸炮制前后阿魏酸的含量測定
2.6.1 線性關系考察 精密量取適量的阿魏酸標準品溶液置于容量瓶中,用甲醇配成系列標準溶液,使濃度分別為1.000、0.100、0.050、0.020、0.010、0.001mg·m L-1,按“2.2”項下色譜條件分別進樣10μL測定。以標準品溶液的質量濃度(X)為橫坐標,峰面積積分值(Y)為縱坐標,制備阿魏酸的標準曲線,得回歸方程為Y=18 361 200.531X+23 738.962(r=0.999 9,n=6)。結果表明,阿魏酸的質量濃度在0.001~1.000mg·m L-1范圍內與峰面積積分值呈良好的線性關系。
2.6.2 精密度試驗 取阿魏酸標準品溶液5μL,按“2.2”項下色譜條件連續進樣6次,測定。結果,RSD=0.57%(n=6),表明儀器精密度良好。
2.6.3 穩定性試驗 取甘肅渭源當歸供試品溶液適量,分別于配制后0、4、8、12、16、24 h進樣測定。結果,RSD=1.13%(n=6),表明供試品溶液中阿魏酸在24 h內穩定。
2.6.4 重復性試驗 取甘肅渭源當歸樣品適量,按“2.4”項下方法平行制備6份供試品溶液,照“2.2”項下色譜條件分別進樣分析。結果,RSD=2.70%(n=6),表明方法重復性良好。
2.6.5 加樣回收率試驗 精密稱取已知含量的甘肅渭源當歸樣品粉末6份,每份0.5 g,分別精密加入相當于含阿魏酸0.4 mg的標準品溶液,混勻,按“2.4”項下方法制備供試品溶液,照“2.2”項下色譜條件進樣測定,計算加樣回收率,結果見表1。
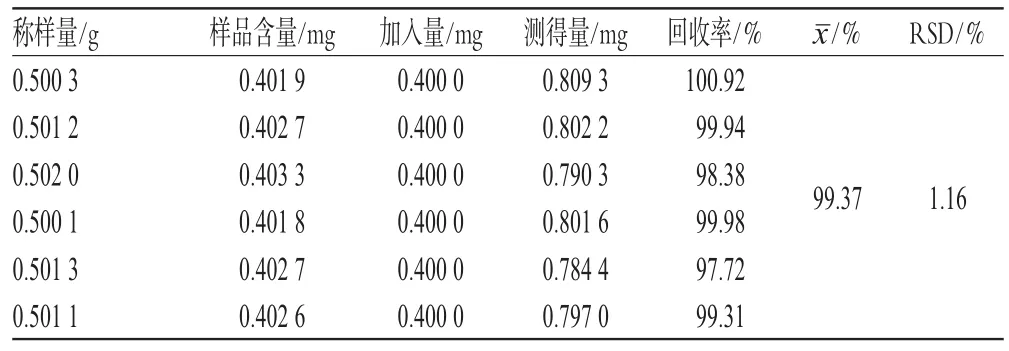
表1 加樣回收率試驗結果(n=6)Tab 1 Resultsof recovery tests(n=6)
2.6.6 炮制前后阿魏酸含量測定 分別精密稱取甘肅漳縣當歸片與酒當歸、甘肅渭源當歸片與酒當歸、甘肅岷縣當歸片與酒當歸樣品適量,分別按“2.4”項下方法制備供試品溶液,照“2.2”項下色譜條件分析。各供試品溶液分別進樣測定3次,以峰面積按標準曲線法計算各樣品中阿魏酸的含量,并計算平均值,結果見表2。
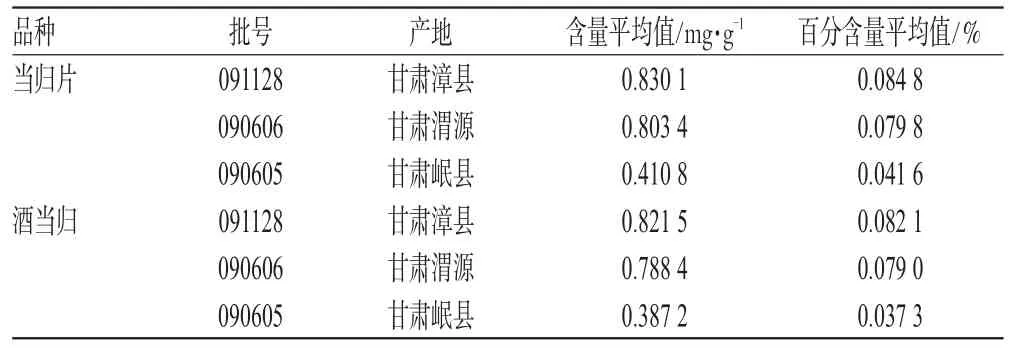
表2 甘肅當歸炮制前后阿魏酸的含量Tab 2 Contents of ferulic acid in crude and processed Gansu A.sinensis
由表2可見,甘肅不同產地當歸中阿魏酸的含量有較大差異,但成分種類和成分間的比例關系基本相似,原因可能為阿魏酸含量受當歸的品種、產地和采收期影響較大。此外,研究還發現甘肅不同產地當歸炮制后阿魏酸含量略有降低。由此可見,在整體特征圖譜相似性良好的情況下,飲片所含有效成分的含量仍然可能存在較大差異。因此,有必要在特征圖譜評價的基礎上增加指標性成分的含量測定,從而全面評價中藥飲片的質量。
3 討論
3.1 樣品提取方法的選擇
本研究對樣品的提取方法進行了考察,以共有峰的峰面積作為評價指標,分別采用加熱回流提取和超聲提取來制備甘肅渭源當歸供試品溶液,提取次數分別考察了1、2、3次,將各樣品按“2.2”項下色譜條件進行液相分析。結果發現,采用不同提取方法、不同提取次數,對8個共有成分的提取率各不相同。考慮到盡量多的提取成分和操作簡單、快速等因素,采用甲醇超聲提取2次,每次30m in作為樣品提取工藝。
3.2 樣品提取溶劑的選擇
本研究分別采用20%、40%、60%、80%和100%甲醇提取甘肅渭源當歸樣品,制備不同提取溶劑的供試品溶液,將各樣品按“2.2”項下色譜條件進行液相分析,以8個共有峰的峰面積為評價指標。結果發現,采用不同提取溶劑對8個共有成分的提取率各不相同。考慮到盡量多的提取成分,采用100%甲醇作為提取溶劑。
3.3 測定波長的選擇
本研究比較了210、230、254、260、280、365 nm等波長下供試品溶液的出峰情況,發現其在254 nm波長處各峰分離好,整體峰形優于其他波長,峰形也好,溶劑干擾少,基線平穩,故選用254 nm作為特征圖譜測定波長。
3.4 流動相的選擇
本研究比較了不同比例甲醇-水、乙腈-水、甲醇-甲酸水溶液、乙腈-甲酸水溶液、甲醇-乙酸水溶液、乙腈-乙酸水溶液等溶劑系統進行梯度洗脫,結果以乙腈-乙酸水溶液系統所得圖譜的峰形較好、分離度高,所以選用乙腈-0.2%乙酸水溶液作為本試驗的流動相系統。
3.5 柱溫和流速的選擇
本研究比較了不同柱溫(25、35、45℃)及不同流速(0.8、1、1.2m L·m in-1)條件下樣品的HPLC特征圖譜。結果發現,柱溫35℃、流速1m L·m in-1時,樣品分離度較好、峰形對稱,能夠滿足分析要求。
3.6 小結
本研究結果表明,特征圖譜技術可以有效進行中藥飲片的鑒別,甘肅不同產地當歸飲片的HPLC特征圖譜差異不大,但是在整體圖譜相似性良好的情況下,藥材所含有效成分的含量仍然可能存在較大差異,如當歸中的主要成分阿魏酸在不同樣品中含量各不相同[6,7]。因此,有必要在特征圖譜評價基礎上增加有效成分的含量測定,從而全面評價中藥飲片的質量,建立更加全面、科學、合理的中藥飲片質量評價體系。
[1]黃偉暉,宋純清.當歸的化學和藥理學研究進展[J].中國中藥雜志,2001,26(3):147.
[2]李 琰,徐麗珍,林 佳,等.不同產地當歸中阿魏酸的含量比較[J].中國藥學雜志,2003,38(11):838.
[3]歐陽曉玫,何英梅,朱俊儒,等.甘肅不同產區當歸藥材的質量考察[J].中國藥師,2007,10(3):206.
[4]宋金春,胡傳芹,劉 紅,等.炮制對當歸藥材有效成分的影響[J].中國藥學雜志,2007,42(14):1 052.
[5]吳 華,李景清,安文源,等.香青蘭藥材HPLC特征圖譜的研究[J].藥物分析雜志,2009,29(7):1 178.
[6]歐陽曉玫,何英梅,朱俊儒,等.甘肅產當歸的商品規格與其阿魏酸含量的相關性初探[J].中國藥事,2005,19(7):423.
[7]王 芳,李 東.當歸的化學及藥理研究進展[J].中國藥房,2003,14(10):630.
