偽威氏按蚊mtDNA-Cytb基因群體遺傳學研究*
2011-08-21 06:51:52湯林華周水森潘嘉云王學忠楊曼尼卓瑪央金
中國人獸共患病學報 2011年12期
劉 茜,武 松,湯林華,周水森,黃 芳,潘嘉云,王學忠,楊曼尼,卓瑪央金
2.中國疾病控制中心寄生蟲病預防控制所,世界衛生組織瘧疾、血吸蟲病和絲蟲病合作中心,上海 200025;
3.云南省寄生蟲病研究所,普洱 665000;
4.西藏自治區林芝地區疾病預防控制中心,林芝 860100
課題組于2007與2010年在西藏瘧疾流行區墨脫縣進行媒介調查發現,該縣的按蚊主要包括多斑按蚊復合體和帶足按蚊,對多斑按蚊復合體進行實驗室種型鑒定發現包括偽威氏按蚊和威氏按蚊[1],其中偽威氏按蚊為絕對優勢蚊種,在偽威氏按蚊中采用巢氏PCR方法檢測出間日瘧原蟲SSU r DNA基因特異序列[2],從而從分子角度判定偽威氏按蚊為該瘧疾流行區的傳瘧媒介。云南省與西藏瘧疾流行區相鄰,偽威氏按蚊在該省分布廣泛。課題組擬采用線粒體細胞色素B基因對西藏、云南和緬甸拉咱市的偽威氏按蚊進行群體遺傳分析,以期為云南省偽威氏按蚊在瘧疾傳播作用方面提供研究思路。
1 材料與方法
1.1 樣本采集 所有按蚊成蚊標本課題組采集于2007年西藏與云南及緬甸現場。采集到的按蚊經過形態學鑒定至多斑按蚊復合體,帶回實驗室采用分子方法進行具體種型鑒定。5個群體共獲偽威氏按蚊共44份樣本用于mt DNA-Cytb基因群體遺傳分析,見表1。

表1 偽威氏按蚊采集地點與編號Tab.1 Anopheles pseudowillimori collection spot and ID
1.2 mt DNA-Cytb基因擴增
1.2.1 基因提取 采用簡易快速DNA提取法[3],即將1~2個蚊腿置于1.5 m L離心管,加入35μL裂解液(10 mmol/L p H8.0 Tirs HCL,1 mmol/L EDTA,1%Nonidet P-40 100μg/m L蛋白酶 K),勻漿;再加入35μL消毒雙蒸水,煮沸5 min;10 000 r/min離心2 min,取上清液置-30℃備用。
1.2.2 擴增體系與條件 采用文獻報道的擴增方案[4],正 向 引 物:5′-GGA CAA ATA TCA TTT TGA GGA GCA ACA G-3′;反向引物為:5′-ATT ACT CCT CCT AGC TTA TTA GGA ATT G-3′。擴增體系為50μL,體系包括10×PCR緩沖液5.0 μL,4.5μL MgCl2(25 mmol/L),1.0μL d NTPs(10 mmol/L),正、反向引物0.8μL(20 mmol/L),2.5 μL redTaq聚合酶(1U/μL),1.5μL DNA 模板,33.9μL dd H2O。反映條件94℃預變性2 min,94℃變性30 s,45℃退火30 s,72℃延伸30 s,共35個循環,72℃孵育8 min。擴增產物送基因公司進行測序。
1.3 數據分析 測序結果以Chromas(Version 2.13)進行核對,必要時手工調整,序列比對采用Clustal X進行,MEGA軟件包統計序列特征,包括堿基含量、轉換和顛換的數目等,并計算序列的差異性(p距離),依據差異性構建鄰接樹(Neighbor Joining Tree,NJ)。將序列數據轉換為ARLEQUIN(Version 3.0)可識別的文件后,統計和計算各群體的基因序列堿基分化參數,置換位點數目、單倍型的類型和在群體中的分布等。單倍型之間的系譜關系由TCS 1.21[5])軟件包構建。群體遺傳結構由ARLEQUIN的AMOVA模塊計算,包括群體間的分化參數Fst(F-statistics)、基因流水平Nm(Nm=1-Fst/4Fst)和群體內和群體間的變異成分。
2 結 果
2.1 擴增情況 受檢偽威氏按蚊DNA標本均擴增出mt DNA-Cytb基因目標片段,擴增片段約為445 bp,所有的擴增產物在瓊脂糖凝膠中觀察,均為單一、清晰的條帶,長度與預期一致。
2.2 偽威氏按蚊mt DNA-Cytb序列特征 測序獲得偽威氏按蚊Cytb基因共44條,比對序列,發現差異極小,經GenBank的BLAST比對,確認該序列片段為特異擴增的Cytb基因。在序列比對后,取其中的420 bp部分片段進行后續分析,結果顯示,堿基置換的位點為26個,其中23處轉換(50.0%),2處顛換(7.69%),1個超變位點(3.85%),共27個單倍型(61.36%),其中有6個單倍型在群體中共享,見表2。該片段的平均AT含量為75.6%,GC含量為24.4%。
2.3 單倍型之間的關系 將本研究得到的偽威氏按蚊Cytb基因的33個單倍型應用TCS1.21軟件包建立95%簡約家系網絡圖(圖1),網絡圖中每個圈代表其中一個單倍型,空白部分是未檢測到的單倍型,在家系網絡圖中各單倍型呈高水平的平行演化,COI_DX02(18)、COI_DX01(12)和 COI_MN16(5)在群體中的分布豐度較高,分別占27.69%、18.46%和7.69%,COI_DX02作為外群的最大可能值0.2125,Cytb_cy15作為外群的最大可能性為0.1925。應用Mega軟件包計算本研究偽威氏按蚊mt DNA-Cytb基因單倍型之間的遺傳距離,并依據遺傳距離構建聚類關系樹(圖2),樹的拓撲關系顯示各單倍型之間的聚類關系沒有明顯的與地理分布呈相關性。
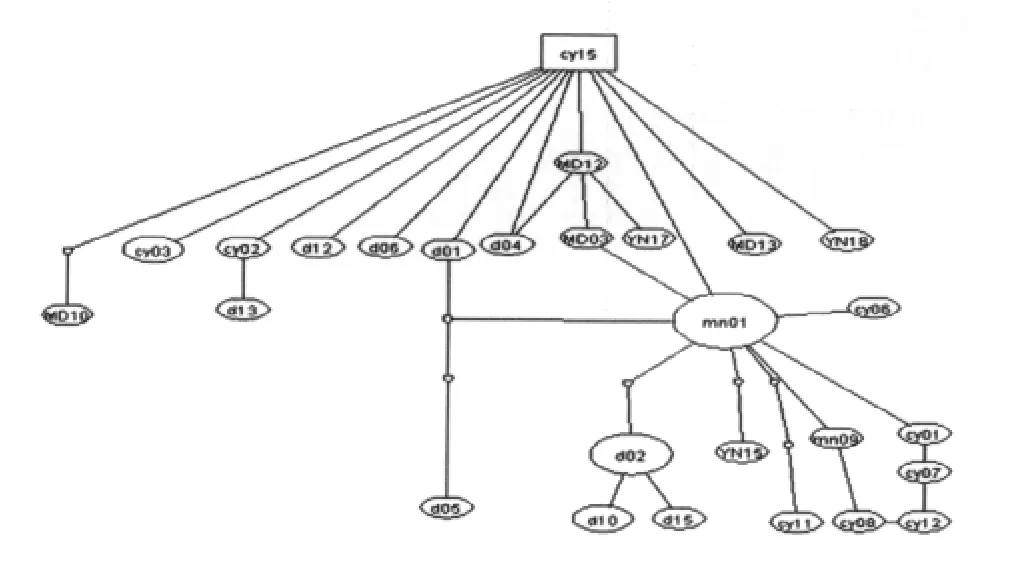
圖1 偽威氏按蚊mtDNA-Cytb基因單倍型的家系網絡圖Fig.1 Family constellation network map of mtDNA-Cytb haplotypes of Anopheles pseudowillimori
2.4 群體遺傳結構 分步AMOVA計算結果顯示,群體內變異絕對大于群體間變異,群體內差異占總差異的98.30%,群體間占1.70%,見表2,所有群體的Fst值為0.016 96,見表3,Nm為4.168,大于1,提示基因流水平可以完全抵御遺傳漂變造成的群體分化,群體間幾乎沒有發生遺傳分化。本研究結果顯示西藏墨脫縣與云南景洪群體間Fst值最大(0.086 50),云南勐臘與緬甸拉咱市、察隅和墨脫群體間的Fst值依次為-0.081 12、-0.003 81和-0.103 03;墨脫與緬甸拉咱市群體間的Fst值為-0.005 12,提示上述群體相互間基因流水平非常高,群體遺傳分化程度很小。
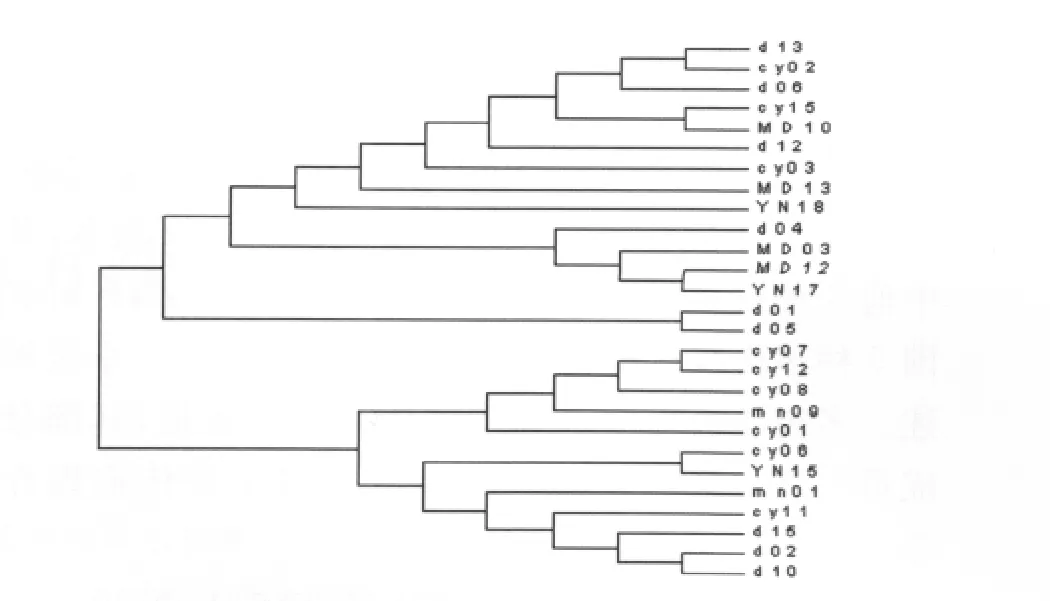
圖2 偽威氏按蚊mtDNA-Cytb基因單倍型之間的聚類關系Fig.2 Cluster relation of mtDNA-Cytb haplotypes of Anopheles pseudowillimori

表2 偽威氏按蚊群體mtDNA-Cytb序列基本數據摘要Tab.2 mtDNA-Cytb gene data of Anopheles pseudowillmori population
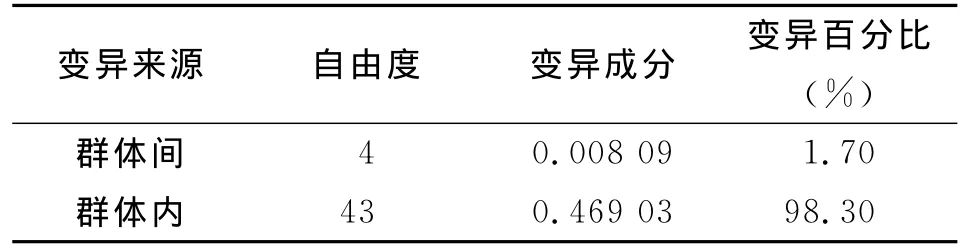
表3 偽威氏按蚊群體mtDNA-Cytb基因AMOVA分析Tab.3 mtDNA-Cytb AMOVA analysis of Anopheles pseudowillmori population
3 討 論
多斑按蚊(Anophelesmaculatus)由Theobald于1901年定名,模式產地為香港,隸屬按蚊屬,塞蚊亞屬,新塞蚊系,主要分布于東洋區(Oriental region),從印度次大陸,經東南亞至中國臺灣。多斑按蚊過去一直被認為是單一蚊種,后經科學家多年研究證實,發現多斑按蚊是由多個形態相似的蚊種組成的復合體,多斑按蚊復合體在我國主要分布于北緯34°以南地區,是山區、半山區及河谷地區的主要蚊種。目前該復合體已經包括9種,Harbach[6-7]最終確立多斑按蚊復合體有9成員種,即多斑按蚊(An.maculatus)、威氏按蚊(An.willmori)、偽威氏按 蚊 (An.pseudowillmori)、塞 沃 按 蚊 (An.sawadwongporni)、達羅毗按蚊(An.dravidicus)、異形按蚊(An.dispar)、諾他按蚊(An.notanandai)、格林按蚊(An.greeni)和多斑按蚊核型 K,其中前5種在我國有分布。姬淑紅[8]和陸寶麟[9]對我國5種多斑按蚊復合體的形態鑒別特征進行系統闡述。多斑按蚊復合體成員種形態非常相似,其部分成員種是馬來西亞、泰國、老撾等國主要傳瘧媒介之一[10-12]。
課題組前期研究在西藏瘧疾流行區墨脫縣發現,偽威氏按蚊為該地的主要傳瘧媒介,云南省與西藏自治區相鄰,董學書[13]發現多斑按蚊復合體中的偽威氏按蚊、威氏按蚊和多斑按蚊在云南省為全省分布,占云南山區、半山區和河谷區按蚊總數的42.68%,其中偽威 氏占 22.44%,多斑 按蚊占9.35%,甚至在云南省某些地區多斑按蚊復合體占捕獲按蚊的比例高達94.6%,為絕對優勢蚊種[14]。并經流行病學調查發現,多斑按蚊與偽威氏按蚊和瘧疾發病關系密切,推測可能為傳瘧媒介;但都因未能發現子孢子感染的直接證據,未能確定其為傳瘧媒介。本研究從群體遺傳學的角度,探討了西藏瘧疾流行區墨脫縣、察隅縣與云南省勐臘、景洪和緬甸拉咱市偽威氏按蚊的群體遺傳分化關系,發現各群體間基因交流頻繁,尚未發生群體間的分化。另群體內變異(98.30%)遠大于群體間變異(1.70%),提示偽威氏按蚊可能曾經歷過近期群體擴張事件,且目前仍處于群體擴張階段,故mt DNA-cytb基因序列的核苷酸變異主要發生在于群體內個體間研究,結果也暗示了云南省的偽威氏按蚊理論上也應具備瘧疾的傳播能力,應該加強云南省偽威氏按蚊傳瘧能力的研究。
[1]武松,潘嘉云,王學忠,等.西藏墨脫縣瘧疾流行區多斑按蚊復合體種型鑒定[J].中國寄生蟲學與寄生蟲病雜志,2008,26(4):286-289.
[2]武松,黃芳,張國慶,等.巢氏PCR判定西藏瘧疾流行區傳瘧媒介[J].中國人獸共患病學報,2010,26(7):648-650,653.
[3]Kambhampati S,Black WC,Rai KS.Random amplified poly morphic DNA of mosquito species and population(Diptera:Culicidae):techniques,statistical analysis,and applications[J].Med Entomology,1992,29(6):939-945.
[4]Beard CB,Hamm DM,Collins FH.The mitochondrial genome of the mosquito Anopheles gambiae:DNA sequence,genome organization,and comparisons with mitochondrial sequences of other insects[J].Insect Mol Biol,1993,2(2):103-124.
[5]Das NG,Bhuyan M,Das SC.Entomological and epoidemiological studies on malaria Rajmahal range,Bihar[J].Lndian J Malariol,2000,37(3/4):88-96.
[6]Harbach RE.The classification of genus Anopheles(Diptera:Culici-dae):a working hypothesis of phylogenetic relationships[J].Bull Entomol Res,2004,94(6):537-553.
[7]Walton C,Somboon P,o’Loughlin SM,et al.Genetic diversity and molecular identification of mosquito species in the Anopheles maculatus group using the ITS2 region of r DNA[J].Infection Genetics and Evolution,2007,7:93-102.
[8]姬淑紅,陸寶麟.中國多斑按蚊類群小記[J].動物分類學報,1991,16(2):224-227.
[9]陸寶麟.中國動物志 昆蟲綱 第九卷 雙翅目 蚊科(下卷)[M].北京:科學出版社,1997:88,92,99,102,106.
[10]Rahman WA,Adanan CR,Abu Hassan A.Species composition of adult Anopheles populations and their breeding habitats in Hulu Perak district,Peninsular Malaysia[J].Southeast Asian J Trop Med Public Health,2002,33(3):547-550.
[11]Kengluecha A,Singhasivanon P,Tiensuwan M,et al.Water quality and breeding habitats of anopheline mosquito in northwestern Thailand[J].Southeast Asian J Trop Med Public Health,2005,36(1):46-53.
[12]Vythilingam I,Phetsouvanh R,Keokenchanh K,et al.The prevalence of Anopheles(Diptera:Culicidae)mosquitoes in Sekong Province,Lao PDR in relation to malaria transmission[J].Trop Med Int Health,2003,8(6):525-535.
[13]董學書.云南多斑按蚊種團的地理分布、生態習性與瘧疾的關系[J].寄生蟲與醫學昆蟲學報,1996,3(2):100-105.
[14]和春桐.云南貢山縣獨龍河谷地帶傳瘧媒介的調查研究[J].現代預防醫學,2008,35(23):4670-4671.
