化藥及生化藥注射劑再注冊審查要求與注意事項
2011-10-30 04:13:46北京市藥品審評中心100053李錚田曉娟周宏佟利家
首都食品與醫藥 2011年2期
北京市藥品審評中心(100053)李錚 田曉娟 周宏 佟利家
根據國家食品藥品監督管理局《關于做好藥品再注冊審查審批工作的補充通知》及北京市藥品監督管理局《關于藥品再注冊審查審批工作的通知》要求,北京市注射劑再注冊工作從2010年9月30日起已集中對到期注射劑品種進行了審查。從已審查的再注冊資料來看,申報資料存在一些問題,須補正資料。與一般品種再注冊資料要求不同,化學藥品注射劑和多組分生化藥注射劑是兩類安全性風險較大的藥品品種,國家食品藥品監督管理局《關于發布化學藥品注射劑和多組分生化藥注射劑基本技術要求的通知》(以下簡稱7號文)明確指出,已經批準注冊的化學藥品注射劑和多組分生化藥注射劑也應參照《技術要求》進行相關研究,并在申報再注冊時提供相關研究資料。現將注射劑再注冊的審查要求與注意事項進行說明與述要。
1 注射劑再注冊的基本原則
1.1 對治療類大容量化學藥品注射劑和多組分生化藥注射劑品種未按7號文要求開展研究工作的,不予再注冊;已開展研究工作并至少完成了無菌工藝驗證和關鍵質量控制項目(如有關物質、熱原等),證明其安全風險可控的,先予以再注冊,在批件中要求1年內完成7號文規定的其余工作,以補充申請形式報國家食品藥品監督管理局。
1.2 除治療類大容量化學藥品注射劑和多組分生化藥注射劑外的品種已按7號文要求提交研究資料的,予以再注冊;尚未按要求提交完整研究資料的,先予以再注冊,在批件中要求1年內完成7號文要求的工作,以補充申請形式報國家食品藥品監督管理局。對其中可最終滅菌工藝F0<8的品種, 應至少提交無菌工藝驗證研究資料,在批件中要求1年內完成其余工作;對可最終滅菌工藝F0<8且未提交無菌工藝驗證研究資料的品種,不予再注冊。
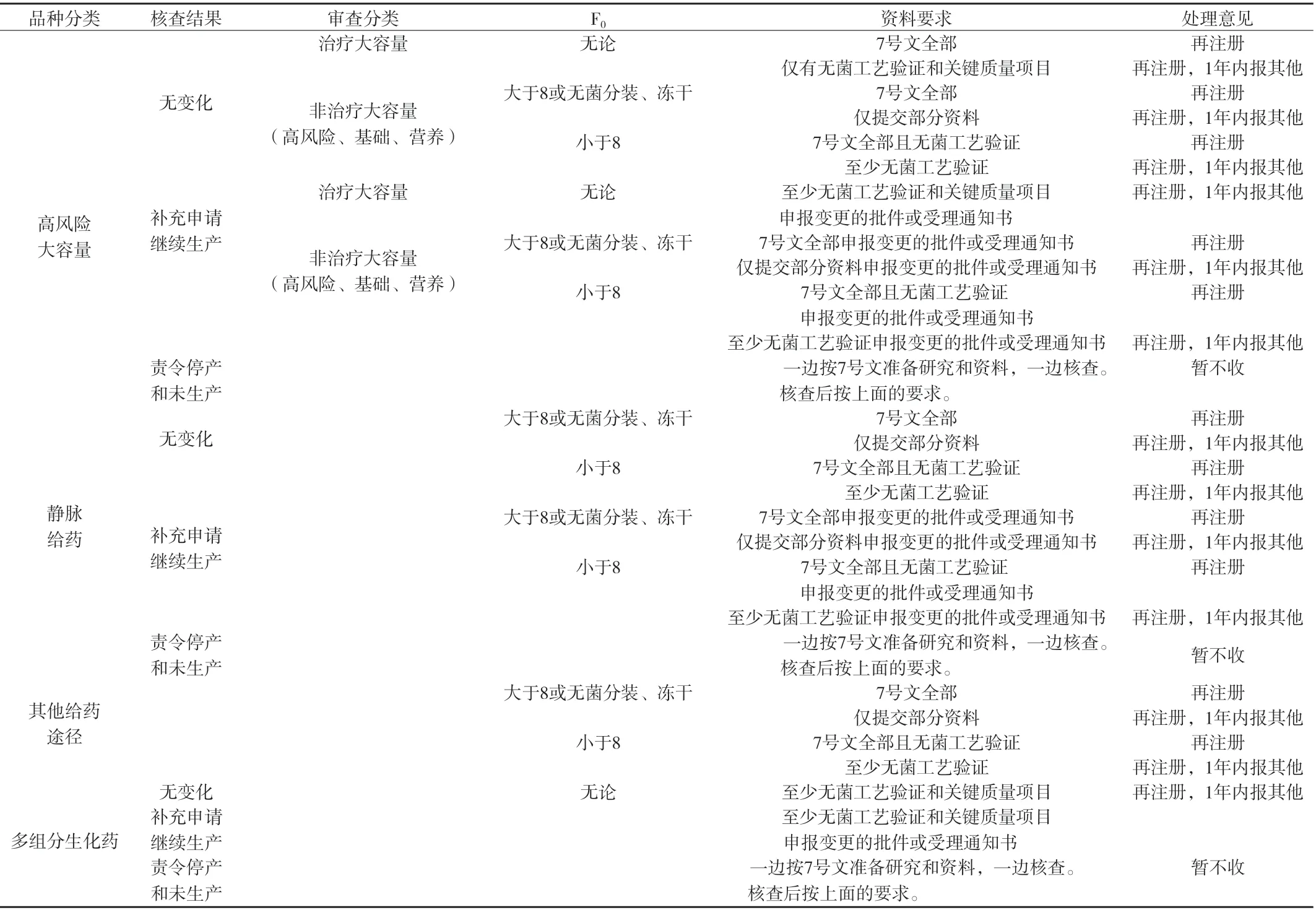
附表 注射劑再注冊審查要點
2 注射劑再注冊的申報資料項目
2.1 證明性文件 藥品批準證明文件、歷次補充申請批件和備案情況公示結果;《藥品生產許可證》復印件包括正本、副本及變更頁的復印件;五年內生產、銷售、抽驗情況總結,對產品不合格情況應當做出說明。
2.2 有下列情形之一的,應當提供相應資料或者說明:藥品批準證明文件或者再注冊批準文件中要求繼續完成工作的,應當提供工作完成后的總結報告,并附相應資料;首次申請再注冊藥品需要進行IV期臨床試驗的,應當提供IV期臨床試驗總結報告;首次申請再注冊藥品有新藥監測期的,應當提供監測情況報告。
2.3 注射劑核查結論 提供有關生產工藝和處方核查結果文件,文件中要求補充研究資料或提出補充申請的,應提交相關報告和補充申請的受理通知書。
2.4 其他資料 按照7號文的要求提供相關研究資料、藥品再注冊申請表、真實性自我保證聲明、授權委托書。
3 注射劑再注冊重點問題
注射劑再注冊申報資料重點是其中的資料4“按照7號文的要求提供相關研究資料”,這部分也正是注射劑與其他品種再注冊區別最大之處。筆者分別從化學藥品注射劑和多組分生化藥注射劑兩個方面予以介紹。
3.1 化學藥品注射劑 劑型選擇的必要性、合理性:以說明形式提供。該品種劑型如為藥典所收載,應在資料中明確指出。如非藥典所收載,應結合該品種自身特點、劑型優勢、上市使用等予以說明。
規格的必要性、合理性:以說明形式提供。該品種規格如為藥典所收載,應在資料中明確指出。如非藥典所收載,應結合該品種臨床使用的用法用量等予以說明。
原輔料質量控制及來源:包括正在使用的化學原料藥的有效批準證明文件、GMP證書;變更或增加原料藥來源的,應當提供批準證明文件或備案情況公示內容。
處方及制備工藝合理性、可行性研究,特別是滅菌工藝的選擇及驗證研究、工藝穩定性研究等:其中應重點說明最終滅菌產品的F0值及無菌控制措施或非最終滅菌產品的無菌保證措施,提交已完成的無菌工藝(無菌生產或滅菌)驗證方案、數據和報告,治療類大容量注射劑品種還應完成關鍵質量控制項目(如有關物質、熱原等)研究。
質量研究及質量標準制訂中,藥品標準均需提交復印件,包括藥典標準。穩定性研究:應提供能夠支持該產品有效期的3批完整的穩定性數據。非臨床安全性、五年內藥品臨床使用情況及不良反應情況。
建議從4個方面予以闡述:對產品處方、生產工藝、工藝中關鍵控制點及其設計進行詳細說明;提供該產品的生產工藝驗證資料(針對該產品專屬生產工藝的驗證方案和驗證報告);提供該產品的工藝回顧性驗證報告(對以往生產該產品的關鍵數據進行匯總分析);提供該產品的無菌工藝驗證資料(無菌生產或滅菌的驗證方案和驗證報告)。
部分驗證工作可結合生產線驗證一并進行:采用終端滅菌工藝的注射劑滅菌工藝驗證主要包括滅菌前微生物污染水平測定、熱穿透試驗、微生物挑戰試驗;采用無菌生產工藝的小容量注射劑和凍干粉針劑的工藝驗證包括設備驗證、環境監測、培養基灌裝驗證、除菌過濾系統適應性驗證;無菌分裝粉針劑的工藝驗證主要為培養基灌裝驗證試驗。
3.2 多組分生化藥注射劑 劑型及規格的合理性:以說明形式提供。該品種劑型及規格如為藥典所收載,應在資料中明確指出,如非藥典所收載,應結合該品種自身特點、劑型優勢、用法用量等予以說明;制備工藝研究:其中應重點說明最終滅菌產品的F0值及無菌控制措施或非最終滅菌產品的無菌保證措施,提交已完成的無菌工藝(無菌生產或滅菌)驗證方案、數據和報告,以及關鍵質量控制項目(如有關物質、熱原等),本部分內容同化學藥品注射劑第4項要求;毒滅活/去除工藝驗證:本部分內容為對多組分生化藥的特殊要求,主要是基于這類品種采用的動物和/或人源性原材料可能引入病毒而制定。本部分內容應針對所采用原材料可能引入的病毒種類、所采用的病毒滅活/去除工藝以及相關驗證報告予以詳細說明;質量研究及穩定性研究:須提交藥品標準復印件。藥理毒理研究:5年內藥品臨床使用情況及不良反應情況。
雖然集中進行的藥品再注冊工作已經基本結束,但后續藥品再注冊申報工作將陸續開展,希望申請人能夠認真學習藥品再注冊相關文件,提高再注冊申報資料質量,順利高效地完成藥品再注冊工作。
猜你喜歡
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
山東冶金(2019年6期)2020-01-06 07:45:54
世界農藥(2019年2期)2019-07-13 05:55:12
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
中國衛生(2016年5期)2016-11-12 13:25:28
銅業工程(2015年4期)2015-12-29 02:48:39
中國衛生(2015年5期)2015-11-08 12:09:48
