鈷鉬基水煤氣變換催化劑及其催化反應工藝
2011-11-09 00:44:00連奕新楊意泉方維平
石油化工 2011年4期
連奕新,楊意泉,方維平
(廈門大學 化學化工學院 醇醚酯化工清潔生產國家工程實驗室,福建 廈門 361005)
特約述評
鈷鉬基水煤氣變換催化劑及其催化反應工藝
連奕新,楊意泉,方維平
(廈門大學 化學化工學院 醇醚酯化工清潔生產國家工程實驗室,福建 廈門 361005)
介紹了國內外CO變換催化劑的發展及其在不同變換工藝中的應用,闡述了Co-Mo基催化劑的催化機理與硫化方法。通過綜合分析中溫變換、中溫變換串聯低溫變換、中溫變換串聯兩段低溫變換和全程低溫變換4種代表性的工藝流程,提出了高效節能型的全程低溫變換工藝將是變換技術的發展方向,開發適應全程低溫變換工藝的高活性和高穩定性的Co-Mo基變換催化劑及其預硫化技術是今后的主要研究目標。
水煤氣變換;鈷鉬基催化劑;全程低溫變換;一氧化碳;氫氣;二氧化碳
自20世紀50年代以來,我國已掌握了以焦炭、無煙煤、焦爐氣、天然氣、油田伴生氣和液態烴多種原料生產氨和尿素的技術,形成了特有的以煤、石油、天然氣為原料,大、中、小生產規模并存的格局。由于石油價格飛漲,基于裝置的經濟性考慮,“輕油”和“重油”型合成氨裝置已不具備市場競爭力,因而以煤為原料制氨等煤化工及其相關技術再度成為研發熱點。煤炭作為我國的主要能源,在化學工業中占有十分重要的地位。近年來,我國煤基合成氨和合成甲醇產業的相關生產技術獲得了全面進步[1-5]。
水煤氣變換反應廣泛應用于合成氨、制氫、合成氣制醇和制烴等催化過程以及城市煤氣中CO含量的調控。近年來,水煤氣變換反應在燃料電池和生物質制氫技術中的應用越來越受重視[6-9]。在水煤氣變換反應中,水蒸氣是反應物,為了降低CO的含量并保證高變換率,水蒸氣往往過量。水蒸氣量的多少是衡量變換工藝能耗的重要標志,因此減少水蒸氣量對工業節能降耗意義重大[10-11]。隨著加壓煤氣化和合成氨生產大型化工藝的出現和發展,相應的變換工藝及其催化劑也有了很大的改進。國內外研究者都在競相研究高效節能型變換工藝,其中最有效的節能措施就是降低變換反應的汽氣比(蒸汽與干煤氣的摩爾比),使變換工序在低汽氣比下操作,因此開發適合低汽氣比的催化劑成為研究重點。特別是各種新型節能流程相繼開發成功,研制適合新工藝的節能型催化劑成為必然[12-13]。
1 變換催化劑的現狀與發展
水煤氣變換反應的研究始于1888年,Fe-Cr基催化劑在1913年研制成功,并在德國BASF公司合成氨廠首次實現工業化應用[10,14]。至今,水煤氣變換反應在工業上應用已有上百年的歷史。傳統的變換催化劑通常指的是Fe-Cr基高溫(350~520℃)變換催化劑,主要以尖晶石結構的Fe3O4為活性相、Cr2O3為主要助劑組成,亦稱為Fe3O4-Cr2O3尖晶石固溶體。以不同制備方法得到的氧化鐵為原料生產的催化劑,其活性亦不同。為提高催化劑性能,部分型號的催化劑中還添加了K2O,CaO,MgO或Al2O3等助劑。Fe-Cr基催化劑具有活性高、熱穩定性好、壽命長和機械強度高等優點,但是此類催化劑在使用中需要大量的過剩水蒸氣,以防止催化劑活性組分Fe3O4被過度還原成金屬鐵或碳化鐵以及在低汽氣比下發生費托合成副反應[15-16]。
從20世紀80年代以來,由于節能流程逐漸成為合成氨工業發展的主流,如凱洛格低能耗流程、ICI-AMV流程及布朗流程等,傳統的高溫變換催化劑已不能滿足合成氨工業發展的要求,研制適合新工藝的節能型催化劑成為必然。為此世界各著名催化劑公司相繼研制出適應低汽氣比的改進型Fe-Cr基高溫變換催化劑,如ICI71-4,SK-201,K6-11,C12-4 等[17-18]。此外作為 Fe-Cr基催化劑中主要穩定劑的Cr2O3,其毒性,特別是致癌性越來越引起了人們的高度關注。因此,研制出無Cr的Fe基或無Cr非Fe基的高溫變換催化劑,降低催化劑在生產、使用過程中的能耗,成為該領域的主要研究方向[19-21]。
隨后開發的活性溫度較低(190~250℃)的Cu-Zn基變換催化劑,主要應用于以天然氣為原料制合成氣的CO低溫變換工藝中,1963年首先在美國的合成氨工業中得到應用[22]。Cu-Zn基變換催化劑的主要活性組分是CuO,添加了ZnO,Al2O3或Cr2O3作為結構助劑,在催化劑制備過程中可形成熱穩定性很高的Zn-Al或Cr-Al尖晶石,從而促進Cu微晶的分散。在過去的幾十年,有大量關于Cu-Zn基低溫變換催化劑制備方法、催化性能和機理的報道,同時Cu-Zn基低溫變換催化劑組分也從最初的 Cu-Zn-Cr發展為 Cu-Zn-Al[23-24]。近年來,對于Cu-Zn低溫變換催化劑的研究主要集中在探討催化反應活性位的微觀組成結構和反應機理以及通過改進其制備方法、添加一些助劑提高其耐熱性[25-27]。但目前的研究結果表明,耐熱性提高幅度有限,而且該類催化劑基本能夠滿足現有生產工藝的要求,所以有關該類催化劑的研究報道逐漸減少。
從工藝方面看,最初使用的Fe-Cr基催化劑由于存在活性溫度較高、水蒸氣消耗量較大等缺點,同時受熱力學平衡的限制,固定床絕熱反應器出口CO體積分數僅能降低到2% ~3%,只能采用銅氨洗工藝脫除剩余的微量CO。如果在甲醇串聯甲烷化流程中采用甲烷化法脫除剩余的微量CO,根據工藝要求,變換氣中CO體積分數需降至0.3%以下,顯然單獨使用Fe-Cr基變換催化劑很難達到要求[19,28]。雖然隨后開發的 Cu-Zn基低溫變換催化劑的活性溫度低,但其耐硫和耐熱性能卻極差,反應溫度高于250℃后活性急劇下降。所以從工藝角度出發,Cu-Zn基低溫變換反應器進口的CO體積分數不能超過5%,以避免由于熱效應造成催化劑床層溫升過大,這一點限制了它的使用范圍[29]。
十幾年來,隨著原料路線多元化制氣技術的發展,要求變換催化劑除具有高的活性外,還需要有良好的抗硫性能。與傳統的Fe-Cr基和Cu-Zn基催化劑相比,Co-Mo基催化劑的特點是只有當活性組分處在硫化狀態下才具有活性,因而不存在硫中毒問題,不需要預脫除原料氣中的硫化物,而且具有操作彈性大、活性高、使用溫區寬(180~500℃)、不易中毒和耐硫等優點。因此許多國家進行了Co-Mo基催化劑的研究開發,并于1978年首次實現工業化生產與應用[30-32]。隨著合成氨和合成甲醇等煤基化工的飛速發展,國內外對Co-Mo基變換催化劑的研究越來越活躍,著重于研制具有活性高、機械強度高、穩定性好、再生能力強、寬汽氣比、寬溫、耐硫、節能、低成本和制備工藝簡單等優點的催化劑。
2 高效耐硫寬溫Co-Mo基變換催化劑
高效耐硫Co-Mo基變換催化劑通常以Ⅵ和Ⅷ族中的某些金屬(如Ni,Co,Mo,W 等)的氧化物或它們的混合物為活性組分,并添加堿金屬(如K和Mg等)為功能性助劑。目前,工業上應用最多的是Co-Mo基寬溫耐硫變換催化劑,在使用前將其轉化為雙金屬硫化態Co-Mo,其中MoS2是催化劑的活性相,Co和 Ni是催化劑的助劑[33-34]。傳統的Co-Mo基變換催化劑的制備方法有混碾法和浸漬法[35]。混碾法是將 Co、Mo(Ni,W)和堿金屬的鹽類水溶液、Al2O3粉、黏結劑等按規定配比混合,經碾壓、造粒或壓片、干燥、焙燒,得到催化劑成品(或將γ-Al2O3粉末浸漬上述鹽類水溶液后擠條成型再經干燥焙燒而成)。浸漬法是將成型的γ-Al2O3等分步浸漬或共浸漬Co、Mo(Ni,W)和堿金屬的鹽類水溶液,經干燥、焙燒,得到催化劑成品。目前得到廣泛應用的負載在γ-Al2O3上的Co-Mo-K/γ-Al2O3催化劑是一種性能優良的耐硫變換催化劑,既可作為中溫耐硫變換催化劑使用,也可作為低溫耐硫變換催化劑使用。
在研究和應用耐硫變換催化劑的過程中發現,以純γ-Al2O3為載體的耐硫變換催化劑在高溫、高壓、高汽氣比的條件下會出現γ-Al2O3的晶相變化、催化劑的活性和強度等指標大幅下降的缺點,進而又研究開發了復合載體,比較典型的載體有Mg-Al尖晶石、Ti-Al尖晶石、Ti改性的Mg-Al尖晶石等。在耐硫變換催化劑的Al2O3載體中加入MgO,特別是形成Mg-Al尖晶石的載體可以改善耐硫變換催化劑的強度、穩定性和寬溫活性。Mg-Al尖晶石因在較高汽氣比和壓力下晶相不變、穩定性好和具有對耐硫變換有利的堿性等而被公認為是理想的耐硫變換催化劑載體[36-38]。特別是以Mg-Al尖晶石為載體的催化劑由于采用了特殊的制備工藝以及添加了新的組分和堿金屬助劑,對高空速、寬汽氣比的適應能力和穩定性高,具有較高的耐油、抗積碳和抗毒物的能力。因此Mg-Al尖晶石作為載體的重要作用越來越引起該領域科研人員的重視。
工業上通常可按是否含有堿金屬K將催化劑分為Co-Mo-K基催化劑和無鉀Co-Mo基催化劑,也可按照適用壓力范圍將催化劑分為兩大類:一類是適用于低壓(小于3.0 MPa)的耐硫低溫變換催化劑Co-Mo-K/γ-Al2O3,這類催化劑制備工藝簡單,低溫活性高,但強度和穩定性差,存在K流失、易反硫化而失活的現象,主要在中小型合成氨廠的中低壓流程中使用;另一類是適用于高壓(3.0~8.0 MPa)、高汽氣比(約1.4)的以Mg-Al尖晶石為載體的耐硫高溫變換催化劑Co-Mo/MgO-Al2O3,這類催化劑具有較好的強度和穩定性,能在高壓、高汽氣比的大中型合成氨廠中的加壓耐硫變換工藝中使用[30]。
國外在Co-Mo基耐硫變換催化劑的研制方面起步較早,目前已報道的耐硫變換催化劑的品種和型號較多,使用較多的工業耐硫變換催化劑[30-35]主要有:(1)1969年德國BASF公司開發成功的K8-11型耐硫變換催化劑,用于重油部分氧化法制合成氣流程和加壓煤氣化制合成氨流程中的CO變換工序,首次在BASF公司路德維希氨廠使用。該型號催化劑的主要特點是以Mg-Al尖晶石為載體,硫化后活性高,耐高水蒸氣分壓,可在高壓下使用,抗毒物能力強,能再生,平均壽命3~5 a;(2)由美國埃克森研究和工程實驗室開發成功的SSK型催化劑,丹麥TopsΦe公司進一步開發,于1974年進行工業應用,主要用于重油氧化法CO變換工藝。該型號催化劑含有較高濃度的K2CO3促進劑,故低溫活性高,同時對毒物不敏感,可耐100 μg/g的氯,存在K流失的缺點,尚無在7.84 MPa下使用的工業數據;(3)美國UCI公司開發的C25-2-02型新一代耐硫變換催化劑,主要用于低壓流程。該型號催化劑的主要特點是含有稀土穩定劑和促進劑,催化劑的結構穩定性好,使用后強度和比表面積保留率高,低溫活性好,抗毒能力強,但未見在7.4 MPa下使用的報道。
“十五”期間,國內建設了多套以煤或渣油為原料的大型合成氨廠,為配合引進裝置所用耐硫變換催化劑的國產化以及滿足國內中小型合成氨廠節能技術改造的需要,我國于20世紀80年代開展了耐硫變換催化劑的研制工作。上海化工研究院從1977年開始進行SB系列耐硫變換催化劑的研究,湖北省化學研究所重點研究了EB系列耐硫變換催化劑的制備及硫化方法,這兩個系列催化劑屬于Co-Mo-K/γ-Al2O3耐硫變換催化劑,主要用于中小型氮肥廠中溫變換串聯低溫變換(簡稱中串低)或全程低溫變換(簡稱全低變)工藝。中國石化齊魯石化公司1988年開始耐硫變換催化劑的研制,并于1992年采用混合法開發出QCS系列的Co-Mo/MgO-Al2O3(或Co-Mo/MgO-TiO2-Al2O3)耐硫變換催化劑,主要用于由煤或渣油為原料高壓氣化生成的含硫原料氣制取合成氣和制氫的大型裝置,目前生產的QCS-1型耐硫變換催化劑已部分替代BASF公司的K8-11型等國外催化劑,在高壓水煤漿氣化流程中應用。廈門大學于1998年開始開發新型水煤氣變換催化劑和變換工藝。采用浸漬法制備了Co-Mo-W-K/γ-Al2O3和Co-Mo/MgO-Al2O3等XH系列多元組分變換催化劑及組合式填裝方法,并于2006年在多家合成氨廠成功投用[39]。
國內外典型的Co-Mo基耐硫變換催化劑的性能及使用條件見表1。
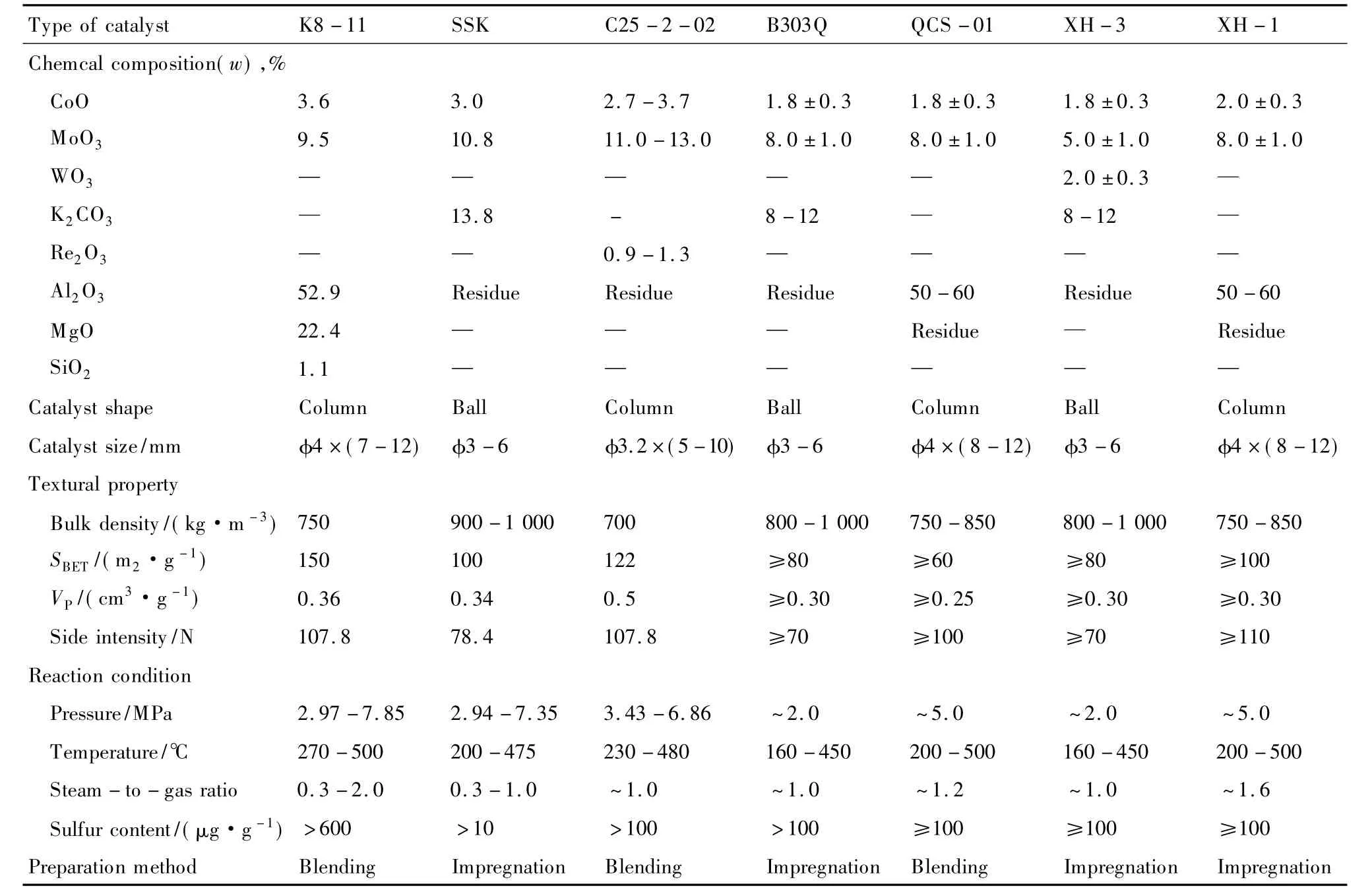
表1 國內外典型的Co-Mo基耐硫變換催化劑的性能及使用條件Table 1 The properties of typical Co-Mo-based sulfur-tolerant water-gas shift catalyst at home and abroad
多年來,Co-Mo基變換催化劑在工業上不斷得到推廣應用,同時催化劑制備方法也不斷改進,積累了許多寶貴的研究成果:
(1)載體與催化劑制備方法的改進[40-43]。采用模板劑或溶膠-凝膠法制備納米粒子載體。相對于傳統的混捏法,浸漬法可以提高活性組分在載體上的分散度,增加活性中心的數量并改善其分布。將多種活性組分一次負載到載體上的共浸漬法比傳統分步浸漬法的浸漬次數少,催化劑活性高。在浸漬液中加入穩定劑,共浸漬后的物料經烘干和焙燒制得催化劑,使用時經硫化處理,其低溫活性好。
(2)催化劑顆粒形態的優化。目前耐硫變換催化劑的顆粒形態一般為圓柱形或球形,若將催化劑制成其他形態,如三葉草和四葉草等,則可以增加顆粒外表面積和提高催化劑床層空隙率,從而提高單位體積催化劑的表觀活性,降低床層流動阻力[44-45]。
(3)對載體進行改性或在催化劑中加入其他助劑[39,46-48]。采用擬薄水鋁石為原料,通過添加造孔劑來調整載體的孔結構,并加入MgO提高載體強度。加入Ti促進催化劑低溫下物理吸附H2S的能力。加入貴金屬或稀土元素,可以使Mo保持較好的分散,提高催化劑的低溫活性。在傳統 Co-Mo-K/γ-Al2O3催化劑中,引入第三活性組分W,提高了催化劑的高溫耐熱性能。
3 Co-Mo基催化劑的催化反應機理
對于CO變換反應機理,最初人們認為催化劑的物理因素起著重要作用,后來在催化劑表面發現了CO與H2O的還原與氧化,提出了氧化-還原機理。隨著研究的進一步深入,人們注意到催化劑表面處于微晶體角、棱、缺陷位置和不規則晶面上的原子和分子具有較大的不飽和性和最大的吸附能力,容易與反應物作用,從而確立了活性中心的概念并提出了吸附機理(締合機理)。后來人們在催化劑上發現了甲酸中間物的形成,又提出了締合機理-伴有中間物的吸附機理。為了更深入地闡明金屬催化的水煤氣反應機理,在羧甲基中間體機理的基礎上又提出了雙功能反應機理。CO和H2O的變換反應,按反應溫度分為高溫變換和低溫變換,而兩者的反應機理又有所不同[49-51]。通過對金屬氧化物及金屬催化生成的甲酸和甲酸根中間體的深入研究,通常認為在金屬或堿性氧化物催化條件下,甲酸根中間體容易分解生成CO2和H2O,變換反應歷程如下[52-53]:
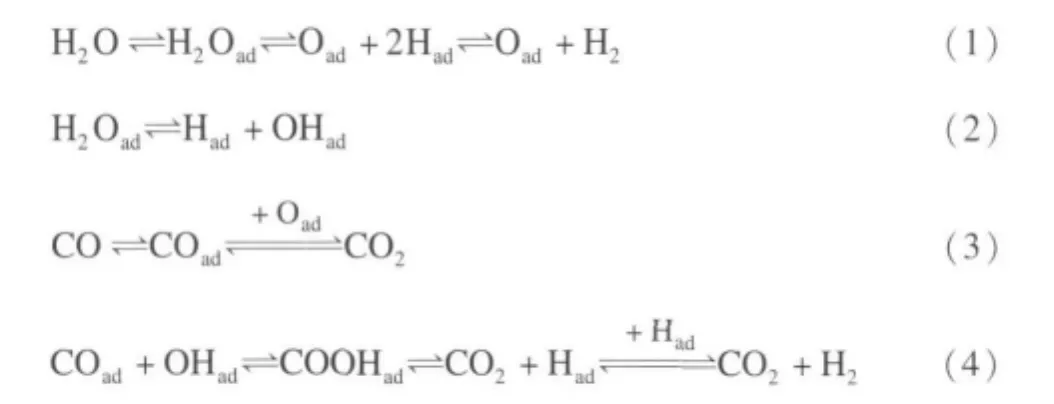
雖然近年來人們對水煤氣變換反應進行了廣泛而深入的研究,由于研究者采用的催化劑、實驗條件、技術手段不盡相同,即使對同類型的機理,也可能會得到不同的表達式。CO變換反應或許不只以一種機理進行,更有可能是幾種機理的綜合作用,同時沒有充足的證據說明在工業條件下哪種機理占優勢。
對于Co-Mo基耐硫變換催化劑,盡管化學成分和含量不同,但主要活性組分是MoO3,且催化劑只有在硫化態下才具有變換活性。范淑蓉等[54]根據對Co-Mo-K/γ-Al2O3催化劑酸性的研究認為,變換反應與Mo5+無關,反應可能按Mo6+-Mo4+之間的雙電子機理進行,而Mo4+為反應中心:
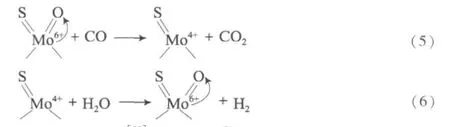
然而,Hou等[55]對MoO3基催化劑的變換反應動力學進行研究發現,原料氣中的硫含量對反應活性起決定性的作用。根據動力學和催化劑的表征認為,在含硫水煤氣變換反應中存在硫化和反硫化的動態平衡,在H2S/H2的還原氣氛中,Mo6+離子還原成Mo5+和Mo4+的共存狀態,變換反應是按照Mo5+-Mo4+之間的氧化-還原機理進行的:
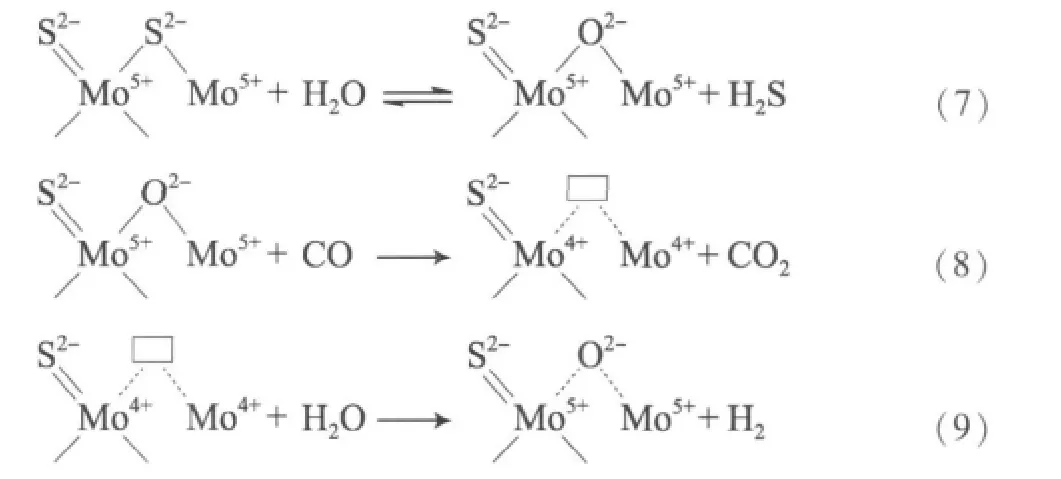
Nikolova等[56]認為在變換反應中,“thio-Mo5+”(S包圍的 Mo)首先與水反應生成“oxysulfo-Mo5+”(O,S包圍的 Mo),然后“oxysulfo-Mo5+”與CO反應生成 CO2,且 Mo5+還原為 Mo4+,然后Mo4+與水反應生成“oxysulfo-Mo5+”。隨著對催化劑的活性與活性物種之間相互關系研究的深入,普遍認為硫化態Mo基催化劑在含硫原料氣中的變換反應按照Mo5+-Mo4+之間的氧化-還原機理進行。同時從反應機理也可看出,由于反硫化反應的存在,會造成H2S的流失,因而在工業生產中原料氣中要維持一定含量的H2S。
4 Co-Mo基催化劑的硫化及器外預硫化
工業上常用的Co-Mo基變換催化劑大多采用Mo,Co,Ni,W 等金屬作為活性組分,并以氧化態形式分散在多孔載體上。Co-Mo基變換催化劑在使用前需要通過活化(硫化)的方法,使氧化態金屬轉變為具有催化活性的硫化態[57-58]。催化劑的硫化技術是催化劑開發和應用的關鍵步驟之一。傳統的硫化方法基本上采用器內氣相硫化工藝,即用原料氣配入液體CS2高溫氫解產生H2S,進行循環硫化或一次放空硫化;個別廠家采用高硫原料氣直接硫化,也有采用固體硫化劑,即在反應器前串聯一個硫化反應器,或將固體硫化劑與催化劑混合裝填在加氫反應器內,通入還原氣在器內實現硫化劑的熱氫解反應產生H2S,H2S再與氧化態Co-Mo催化劑發生硫化反應。
器內氣相硫化法的基本過程包括:催化劑的升溫階段(常溫~220℃),主要是脫除催化劑中的物理水;催化劑的硫化階段(220~350℃),主要是催化劑中表層的活性組分進行充分硫化;強化硫化階段(350~420℃),主要通過升高硫化溫度促使催化劑完全硫化;整個硫化時間一般在50 h以上。采用器內氣相硫化法的優點在于可以減少硫化劑損失,硫化較為均勻,但存在以下缺點:需要專用的預硫化管路和設備;硫化時間較長;硫化過程中催化劑床層溫度波動大;硫化劑均為有毒有害物質;易造成催化劑硫化不完全。在催化劑制備過程中,由于高溫焙燒分解生成的氧化物與載體之間存在強烈的相互作用,往往導致硫化不完全[59-61]。
近年來,具有特有優點的器外預硫化技術得到普遍重視[59-61]。器外預硫化較常采用的方法是:將活性金屬為氧化態的催化劑先與硫化劑混合,再裝入反應器中,開工時只需通入氫氣或同時通入含氫原料氣,隨后升溫即可完成硫化。常用的硫化劑為單質硫、無機或有機多硫化物。器外預硫化技術的優點在于:開工時間短;活化期間催化劑床層溫度穩定,容易操作;現場不需要再準備硫化劑,減少對環境的污染;不需要專用的預硫化設施;催化劑能夠充分硫化;催化劑的活性較器內氣相硫化的高或相當。目前器外預硫化技術在國外已經商業化,特別是在加氫脫硫工藝中;國內也進行了初步應用。因此,深入研究Co-Mo基催化劑的預硫化方法對開發高活性催化劑有重要意義。
Lian等[62]采用(NH4)2S溶液為預硫化劑制備了Co-Mo-S/MgO-Al2O3預硫化催化劑,研究發現,以H2S/H2/N2混合氣進行器內氣相硫化時,按照先硫化再還原的過程進行,也就是低溫下先發生氧硫交換,MoO3前體中的端基O2-被S2-取代形成了Mo—S鍵,而氧硫交換繼續進行,促使橋式S2-2和Mo5+中心形成;升高溫度后Mo5+物種開始還原形成Mo4+存在于MoS2物種中。而在器外浸漬硫化銨的催化劑,先在催化劑表面形成含硫金屬化合物前體,該含硫金屬化合物前體在合成氣條件下經歷了熱分解和還原過程[63-64]:(NH4)2MoS4(200 ~300℃)→MoS3(350~400℃)→MoS3(無定形態)(>500℃)→ MoS2(晶態)。器外預硫化與傳統器內H2S硫化具有不同的活化機理。他們同時指出在預硫化時,Mo的化合物同樣也會發生氧硫交換,在催化劑表面生成(NH4)2MoS4或(NH4)2MoOxSy,同時也可能存在部分其他硫化產物,如 MoOS,MoOS2,MoO2S等,甚至會出現組成和結構更為復雜的成分。在催化劑表面形成的硫化產物的表面上,硫是同時與Mo4+和Mo6+化合的,不僅僅存在于MoS2中。硫化態前體制備的MoS2/MgO-Al2O3催化劑本身已是硫化態,降低了活性組分與載體的相互作用,有利于形成 MoS2和CoMoS物種,提高硫化度,因而具有較高的變換活性和耐熱性能。
先進的器外預硫化技術能克服傳統器內氣相硫化的缺點,使催化劑保持最佳的活性和穩定性,提高選擇性,延長使用壽命。同時預硫化催化劑經過鈍化處理后,可有效降低催化劑的自熱性及SO2的生成,催化劑的固硫性能好,從而使催化劑保持活性和穩定性[65-66]。因此,深入研究CO變換催化劑的預硫化及鈍化方法,對開發高活性、可工業化的預硫化CO變換催化劑有重大的現實意義。
5 高效節能變換工藝及其發展
煤氣化是煤化工的龍頭和基礎,很大程度上影響煤化工的效率、成本和發展。根據原料種類和造氣方法的不同,以煤為原料氣的化工工藝可大致分為以下幾種流程:常壓固定層間歇氣化、水煤漿加壓氣化、碎煤加壓氣化、粉煤加壓氣化。制取水煤氣所用原料和氣化工藝不同,水煤氣中CO含量也不盡相同,例如,以煤或焦炭為原料,采用間歇式固定層煤氣發生爐制得的水煤氣中CO體積分數為25% ~30%;以天然氣為原料,采用水蒸氣轉化法制得的水煤氣中CO體積分數為13% ~18%;以重油或渣油為原料,經部分氧化制得的水煤氣中CO體積分數為45%~50%;由水煤漿氣化制得的半水煤氣中CO體積分數為40% ~45%;粉煤加壓氣化制得的水煤氣中CO體積分數高達60%。同時,不同的原料所制工藝氣中硫的含量也有很大差異,因而選用的變換催化劑和變換工藝也有所不同[67-68]。然而,不管采用哪一種原料,合成氨生產工序都必須經過煤氣化、水煤氣變換、合成氣凈化、合成氣壓縮及合成過程,只不過在具體細節上有所不同,并隨著合成氨技術的發展而不斷演化[69]。
發達國家的變換工藝基本上都使用寬溫型Co-Mo基催化劑取代傳統的Fe-Cr基中溫變換催化劑,而且國外的合成氨裝置規模一般比較大,不管是原料、操作壓力的選擇、工藝流程還是催化劑與我國的中小型氮肥廠大不相同。我國氮肥企業開始于20世紀60年代,由常壓變換改為加壓變換,每噸氨水蒸氣消耗高達1 200 kg;70年代高活性催化劑的普遍應用使每噸氨水蒸氣消耗降到900 kg左右。隨后在80年代我國開始引進國外第二代煤氣化技術,采用加壓連續氣化制取合成氨原料氣,氣化壓力既有較低的2.8 MPa,也有較高的6.5 MPa。加壓連續氣化制取的工藝氣不僅含有大量的水蒸氣,而且H2S和有機硫含量較高,其中,水蒸氣可以用來進行變換反應,H2S可以保證Co-Mo基耐硫中溫變換催化劑處于活性態,相應的CO變換反應大都要求采用加壓耐硫變換工藝。Co-Mo基耐硫催化劑的研制成功使變換工藝得到重大變革,80年代由中溫變換(簡稱中變)工藝改為中串低工藝,90年代發展了全低變工藝;為了克服全低變工藝不能長周期運行的問題,又開發了中溫變換串聯兩段低溫變換(簡稱中低低)工藝。
目前,國內中小型合成氨廠大都采用以煤為原料、間歇制氣的生產工藝,與之配套的后續凈化工藝主要有銅氨洗和甲烷化兩種流程。銅氨洗凈化流程要求變換氣中CO體積分數低于3.0%,采用的變換工藝主要有中變工藝、中串低工藝、中低低工藝和全低變工藝等;而甲烷化凈化流程則要求變換氣中的CO體積分數小于0.3%,采用的變換工藝主要有兩次脫碳的Fe-Cr基中變催化劑串聯Cu-Zn基低溫變換(簡稱低變)催化劑的變換工藝、Fe-Cr基中變催化劑串聯二段Co-Mo基耐硫低變催化劑的中低低深度變換工藝、全部使用Co-Mo基耐硫低變催化劑的全低變深度變換工藝[70-72]。
5.1 與銅氨洗凈化流程相配套的變換工藝
5.1.1 中變工藝
中變工藝一般是一個變換爐,爐內裝填Fe-Cr基催化劑,分兩段或三段裝填,半水煤氣從上到下依次通過各段催化劑完成變換過程。工藝指標:入爐半水煤氣溫度330~350℃、熱點溫度480~510℃、出爐變換氣溫度400~420℃、出口變換氣中CO體積分數3% ~5%、入爐汽氣比0.7~0.8、每噸氨水蒸氣消耗1 000~1 200 kg。催化劑的終態溫度較高,水蒸氣消耗很大。目前采用中變工藝的合成氨廠逐漸減少。
5.1.2 中串低工藝
Fe-Cr基中變催化劑后再串聯Co-Mo基耐硫低變催化劑,中變氣經熱交換后入低變爐。低變催化劑可放在中變爐最后一段,也可另設一低變爐串聯在中變爐之后。中串低工藝流程的設置可分為爐內串低變增濕流程、爐內串低變調溫水加流程、爐外中串低調溫水加流程及爐外中串低增濕流程。工藝指標:中變爐入口溫度320~340℃、熱點溫度460~480℃、中變爐出口溫度380~400℃;低變爐入口溫度200~220℃、低變爐出口溫度240~260℃;中變爐出口CO體積分數5% ~6%、低變爐出口CO體積分數1% ~2%;入中變爐汽氣比0.5~0.6、每噸氨水蒸氣消耗500~600 kg。該工藝不僅低變催化劑的入口溫度可降低100℃以上、每噸氨水蒸氣消耗顯著降低,取得了明顯的節能效果,而且工藝改造較為簡單,投資少,因此該工藝在低變工藝發展初期得到國內中小型合成氨廠的普遍采用。但是中串低流程存在明顯的不足,主要是中變二、三段催化劑利用率很低,總的催化劑用量大,而生產能力卻不能得到充分發揮。
5.1.3 中低低工藝
中低低變換工藝就是在中串低的基礎上再串聯一段低變段或低變爐。兩個低變爐(段)之間要有降溫措施,用水冷激或水加熱器降溫均可。一般有兩種流程:中變增濕的中低低流程和調溫水加的中低低流程。工藝指標:中變爐入口溫度300~320℃、熱點溫度430~440℃、中變爐出口溫度360~380℃;低變爐每段入口溫度200~220℃、出口溫度240~260℃;中變爐出口CO體積分數10% ~12%、低變爐出口CO體積分數1% ~2%、入爐汽氣比0.4~0.5、每噸氨水蒸氣消耗約400~500 kg。該工藝與中串低工藝本質上沒有區別,由于多了低變爐(段),反應終態溫度比中串低工藝低,因而汽氣比有所下降,節能效果更佳,特別適合老變換系統擴大節能能力的改造。但中低低流程由于中變催化劑減少,一旦漏氧、熱交換器或水加熱器泄漏,第一低變催化劑極易中毒,因而要求催化劑有較強的抗毒性能,否則嚴重影響使用效果。同時由于第二低變段反應的汽氣比、CO含量及反應溫度都較低,要求第二低變段采用活性更高的催化劑,否則節汽效果不理想。
5.1.4 全低變工藝
全低變工藝是在中串低工藝基礎上發展起來的全新變換工藝,變換爐各段采用Co-Mo基耐硫低變催化劑。全低變工藝的流程有兩種:調溫水加的全低變流程、增濕的全低變流程。工藝指標:入爐溫度200~220℃、熱點溫度380~400℃左右、出爐溫度250~260℃;變換爐出口CO體積分數1% ~2%、入爐汽氣比0.3~0.4、每噸氨水蒸氣消耗200~300 kg。相比而言,全低變工藝的節能效果最為明顯,但全低變工藝反應溫度低,難以分解煤氣帶入的焦油和潤滑油,它們容易覆蓋在催化劑表面,使一段Co-Mo基催化劑老化、失活,一段催化劑床層阻力容易升高,導致全低變工藝的工業應用成功率不高,未取得預期效果,相當一部分裝置返回到中串低流程。
5.2 與甲烷化凈化流程相配套的變換工藝
5.2.1 Fe-Cr基中變催化劑串聯Cu-Zn基低變催化劑的變換工藝
Fe-Cr基中變催化劑工藝與銅氨洗凈化流程中的Fe-Cr基中變催化劑工藝基本一致,但以煤為原料制半水煤氣的工廠在Fe-Cr基中變催化劑后無法直接串聯Cu-Zn基低變催化劑,需經二次脫碳、脫硫后再進入Cu-Zn基低變催化劑段。工藝指標:中變爐入口溫度300~350℃、熱點溫度480~500℃、出口溫度400~420℃、CO體積分數3% ~5%;低變爐入口溫度190~200℃、出口溫度230~250℃、出口CO體積分數小于0.3%;中變爐入爐汽氣比0.8~0.9、低變爐入爐汽氣比0.2~0.3、每噸氨水蒸氣消耗1 100~1 300 kg。
5.2.2 中低低深度變換工藝
中低低深度變換工藝一般多采用二段Fe-Cr基中變催化劑串聯二段Co-Mo基耐硫低變催化劑。工藝指標:中變爐入口溫度300~320℃、熱點溫度460~480℃、出口溫度380~400℃、出口CO體積分數5% ~6%;低變爐一段入口溫度200~220℃、二段入口溫度180~200℃、出口溫度200~220℃、出口CO體積分數小于0.3%;中變爐入爐汽氣比0.5~0.6、每噸氨水蒸氣消耗約700~800 kg。
5.2.3 全低變深度變換工藝
全低變深度變換工藝與上述全低變工藝相似,一般采用三段Co-Mo基耐硫低變催化劑。工藝指標:一段入口溫度200~220℃、熱點溫度380~400℃、二段入口溫度200~220℃、三段入口溫度180~200℃、出口溫度210~230℃左右;變換出口CO體積分數小于0.3%、入爐汽氣比0.5~0.6、每噸氨水蒸氣消耗600~700 kg。
變換工藝的變革和進步始終圍繞著催化劑、變換爐和變換氣熱能回收方式進行。變換反應中汽氣比的高低直接影響變換工藝的水蒸氣消耗,汽氣比越低,過剩水蒸氣量越小,因而全低變工藝的水蒸氣消耗最低。同時在合成氨廠中,對變換氣的高位能與潛熱的綜合利用對節能降耗起著重要的作用。早期,中變或中串低流程一般配置飽和熱水塔進行熱量回收,典型流程見圖1。
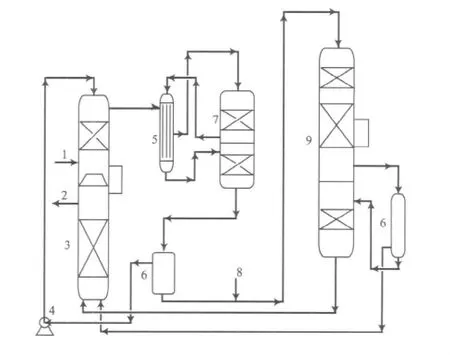
圖1 帶飽和熱水塔的中串低流程Fig.1 Simplified process flow diagram of medium-low temperature shift process with saturation tower.
隨著低變技術的逐步成熟和完善,變換氣中過量水蒸氣已經很少,可以回收的熱量也逐漸減少,因而開始采用無飽和熱水塔變換工藝及副產中(低)壓水蒸氣技術,典型流程見圖2。
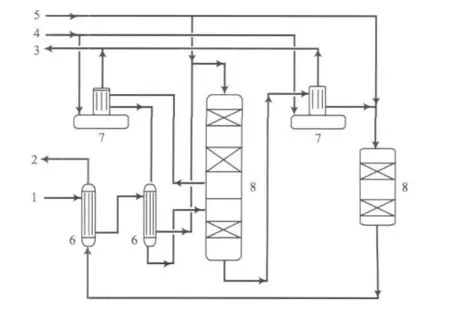
圖2 無飽和熱水塔的全低變流程Fig.2 Simplified process flow diagram of total low temperature shift process without saturation tower.
綜合來說,變換工藝經歷了常壓變換到加壓變換,在加壓的基礎上又經歷了中變、中串低、中低低和全低變幾個過程,每次技術革新,節能效果都明顯改進。但并不是每一個工藝都完美無缺,它們都有其局限性,但總體來說節能效果越來越好。隨著變換催化劑性能的不斷改進以及新工藝和新設備的不斷出現,將會促進熱能的綜合利用,變換系統的水蒸氣消耗會大大降低。中變逐漸會被低變所代替,全低變技術將會成為CO變換工藝的主流方向。
6 結語
為了滿足節能減排的需要,世界上出現了多種降低合成氨能耗的新工藝,特別是合成氨大型化、制氣的加壓化技術的發展,高活性低能耗變換催化劑及其催化工藝的開發越來越受到人們的重視,高效率節能型變換工藝將是今后變換工藝的發展方向。
變換工藝的核心在于催化劑,催化劑的性能直接影響到變換工藝的效能。通過綜合分析中變、中串低、中低低和全低變4種代表性變換工藝及其催化劑的特點可知,目前在中小型合成氨廠能穩定運行的流程為中低低變換工藝,節能效果最佳的流程為全低變工藝,但其所用一段全低變催化劑的性能尚需改進。因此開發出能適應中低壓變換反應爐一段變換的耐硫變換催化劑,對我國中小型合成氨廠進行節能改造具重要意義。同時深入研究CO變換催化劑的預硫化及其鈍化方法,開發高活性、可工業化的預硫化CO變換催化劑將是另一重要課題。
[1]Antonio A,Sigurd S.Control Structure Design for the Ammonia Synthesis Process[J].Comput Chem Eng,2008,32(12):2920-2932.
[2]Deng Xue,Wang Hewu,Huang Haiyan,et al.Hydrogen Flow Chart in China[J].Int J Hydrogen Energy,2010,35(13):6475-6481.
[3]Longwell J P,Rubin E S,Wilson J.Coal:Energy for the Future[J].Progr Energ Combust Sci,1995,21(4):269-360.
[4]於子方.90年代合成氨工藝技術的新進展[J].石油化工動態,1997,5(5):33-38.
[5]陳元春,金小娟.我國煤化工產業發展狀況評述[J].煤炭工程,2009,(5):90-92.
[6]Dong Ju Moon.Low Temperature WGS Catalysts for Hydrogen Station and Fuel Processor Applications[J].Catal Surv Asia,2009,13(3):191-204.
[7]李速延,周曉奇.CO變換催化劑的研究進展[J].煤化工,
2007,129(2):31-34.
[8]Ruettinger W,Ilinich O,Farrauto R J.A New Generation of Water Gas Shift Catalysts for Fuel Cell Applications[J].J Power Sources,2003,118(1-2):61-65.
[9]Galvita V,Schr?der T,Munder B,et al.Production of Hydrogen with Low COx-Content for PEM Fuel Cells by Cyclic Water Gas Shift Reactor[J].Int J Hydrogen Energy,2008,33(4):1354-1360.
[10]Laniecki M,Malecka-Grycz M,Domka F.Water-Gas Shift Reaction over Sulfided Molybdenum Catalysts:Ⅰ.Alumina Titania and Zirconia-Supported Catalysts[J].Appl Catal,A,2000,196(2):293-303.
[11]Baier T,Kolb G.Temperature Control of the Water Gas Shift Reaction in Microstructured Reactors[J].Chem Eng Sci,2007,62(17):4602-4611.
[12]楊玲菲,寧平,田森林,等.低汽氣比節能變換催化劑研究進展[J].化工進展,2009,28(S1):49-53.
[13]Pen? M A,Gómez J P,Fierro J L G.New Catalytic Routes for Syngas and Hydrogen Production[J].Appl Catal,A,1996,144(1-2):7-57.
[14]Liu Quansheng,Ma Wenping,He Runxia,et al.Reaction and Characterization Studies of an Industrial Cr-Free Iron-Based Catalyst for High-Temperature Water Gas Shift Reaction[J].Catal Today,2005,106(1-4):52-56.
[15]Martos C,Dufour J,Ruiz A.Synthesis of Fe3O4-Based Catalysts for the High-Temperature Water Gas Shift Reaction[J].Int J Hydrogen Energy,2009,34(10):4475-4481.
[16]呂待清.鐵基高溫變換催化劑熱力學穩定性分析[J].化肥工業,2006,33(5):25-29.
[17]歐曉佳,程極源.鐵鉻系高(中)溫變換催化劑的研究現狀[J]. 化學研究與應用,1999,11(2):126-131.
[18]Lei Yun,Cant N W,Trimm D L.The Origin of Rhodium Promotion of Fe3O4-Cr2O3Catalysts for the High-Temperature Water-Gas Shift Reaction[J].J Catal,2006,239(1):227-236.
[19]華南平,楊平,杜玉扣.CO高溫變換催化劑發展趨勢[J].小氮肥設計技術,2005,26(4):1-4.
[20]Lee Joon Yeob,Lee Dae-Won,Lee Kwan-Young,et al.Cr-Free Fe-Based Metal Oxide Catalysts for High Temperature Water Gas Shift Reaction of Fuel Processor Using LPG[J].Catal Today,2009,146(1-2):260-264.
[21]Gawade P,Mirkelamoglu B,Tan B,et al.Cr-Free Fe-Based Water-Gas Shift Catalysts Prepared Through Propylene Oxide-Assisted Sol-Gel Technique[J].J Mol Catal A:Chem,2010,321(1-2):61-70.
[22]Lima A A G,Nele M,Moreno E L,et al.Composition Effects on the Activity of Cu-Zn-Al Based Catalysts for the Water Gas Shift Reaction:A Statistical Approach[J].Appl Catal,A,1998,171(1):31-43.
[23]李速延,周曉奇.CO變換催化劑的研究進展[J].煤化工,2007,129(2):31-34.
[24]Wu Jingang,Saito Masahiro.Improvement of Stability of a Cu/Zn/Al Catalyst for the CO Shift Reaction[J].J Catal,2000,195(2):420-422.
[25]李選志,韋孫昌.銅基一氧化碳低溫變換催化劑的研究進展[J].大氮肥,2006,29(1):69-72.
[26]Koryabkina N A,Phatak A A,Ruettinger W F,et al.Determination of Kinetic Parameters for the Water-Gas Shift Reaction on Copper Catalysts Under Realistic Conditions for Fuel Cell Applications[J].J Catal,2003,217(1) :233-239.
[27]Yahiro Hidenori,Nakaya Kenta,Yamamoto Tetsuya,et al.Effect of Calcination Temperature on the Catalytic Activity of Copper Supported on γ-Alumina for the Water-Gas-Shift Reaction[J].Catal Commun,2006,7(4):228-231.
[28]Panagiotopoulou P,Kondarides D I.A Comparative Study of the Water-Gas Shift Activity of Pt Catalysts Supported on Single(MOx)and Composite(MOx/Al2O3,MOx/TiO2)Metal Oxide Carriers[J].Catal Today,2007,127(2)319-329.
[29]劉全生,張前程,馬文平,等.變換催化劑研究進展[J].化學進展,2005,17(3):389-398.
[30]Nagai Masatoshi,Matsuda Kenji.Low-Temperature Water-Gas Shift Reaction over Cobalt-Molybdenum Carbide Catalyst[J].J Catal,2006,238(2):489-496.
[31]Laniecki M,Ignacik M.Water-Gas Shift Reaction over Sulfided Molybdenum Catalysts Supported on TiO2-ZrO2Mixed Oxides Support Characterization and Catalytic Activity[J].Catal Today,2006,116(2):400-407.
[32]Chianelli R R,Berhault G,Torres B.Unsupported Transition Metal Sulfide Catalysts:100 Years of Science and Application[J].Catal Today,2009,147(3-4) :275-286.
[33]Hakkarainen R,Salmi T,Keiski R L.Water-Gas Shift Reaction on a Cobalt-Molybdenum Oxide Catalyst[J].Appl Catal,A,1993,99(2) :195-215.
[34]Mohamed M M,Salama T M,Othman A I,et al.Low Temperature Water-Gas Shift Reaction on Cerium Containing Mordenites Prepared by Different Methods[J].Appl Catal,A,2005,279(1-2):23-33.
[35]石自更,王峰.K8-11和QDB-04型耐硫變換催化劑在殼牌粉煤氣化制甲醇裝置的應用[J].化肥設計,,2009,47(2):33-36.
[36]Kirszensztejn P,Przekop R,Szymkowiak A,et al.Preparation of MgO-Al2O3Binary Gel System with Mesoporous Structure[J].Microporous Mesoporous Mater,2006,89(1-3):150-157.
[37]袁素珺,張青紅,李耀剛,等.高比表面積的鎂鋁復合氧化物納米薄片的制備及其吸附性能[J].硅酸鹽通報,2009,28(4):636-640.
[38]Sarkar R,Bannerjee G.Effect of Addition of TiO2on Reaction Sintered MgO-Al2O3Spinels[J].J Eur Ceram Soc,2000,20(12):2133-2141.
[39]Wang Huifang,Lian Yixin,Li Yinong,et al.W-Promoted Co-Mo-K/γ-Al2O3Catalysts for Water-Gas Shift Reaction[J].Catal Commun,2009,10(14):1864-1867.
[40]李小定,陳勁松.鈷鉬系一氧化碳變換催化劑的制備方法:中國,90102336[P].1993-10-27.
[41]Nickolov R N,Edreva-Kardjieva R M,Kafedjiysky V J,et al.Effect of the Order of Potassium Introduction on the Texture and Activity of Mo/Al2O3Catalysts in Water Gas Shift Reaction[J].Appl Catal,A,2000,190(1):191-196.
[42]Nagai M,Zahidul Md A,Matsuda K.Nano-Structured Nickel-Molybdenum Carbide Catalyst for Low-Temperature Water-Gas Shift Reaction[J].Appl Catal,A,2006,313(2):137-145.
[43]連奕新,王會芳,張元華,等.焙燒溫度對鎂鋁復合氧化物載體性能的影響[J]. 石油化工,2009,38(6):622-628.
[44]縱秋云,田兆明,譚永放,等.QCS-4耐硫變換催化劑的研制[J]. 齊魯石油化工,1998,26(1):14-18.
[45]Mellor J R,Coville N J,Sofianos A C,et al.Raney Copper Catalysts for the Water-Gas Shift Reaction:Ⅰ.Preparation,Activity and Stability[J].Appl Catal,A,1997,164(1-2):171-183.
[46]Nagai M,Zahidul Md A,Kunisaki Y,et al.Water-Gas Shift Reactions on Potassium-and Zirconium-Promoted Cobalt Molybdenum Carbide Catalysts[J].Appl Catal,A,2010,383(1-2):58-65.
[47]Panagiotopoulou P,Kondarides D I.Effect of Morphological Characteristics of TiO2-Supported Noble Metal Catalysts on Their Activity for the Water-Gas Shift Reaction[J].J Catal,2004,225(2):327-336.
[48]Boccuzzi F,Chiorino A,Manzoli M,et al.Gold,Silver and Copper Catalysts Supported on TiO2for Pure Hydrogen Production[J].Catal Today,2002,75(1):169-175.
[49]郭曉勇.負載型金屬納米催化劑的制備及其對水煤氣的催化變換作用[D].蘇州:蘇州大學,2007.
[50]Rhodes C,Hutchings G J,Ward A M.Water-Gas Shift Reaction:Finding the Mechanistic Boundary[J].Catal Today,1995,23(1):43-58.
[51]Natesakhawat S, Wang Xueqin, Zhang Lingzhi, etal.Development of Chromium-Free Iron-Based Catalysts for High-Temperature Water-Gas Shift Reaction[J].J Mol Catal A:Chem,2006,260(1-2):82-94.
[52]Shi Xuerong,Wang Shengguang,Hua Jia,et al.Density Functional Theory Study on Water-Gas-Shift Reaction over Molybdenum Disulfide[J].Appl Catal,A,2009,365(1):62-70.
[53]Li Yumin,Wang Rejie,Chang Liu.Study of Reactions over Sulfide Catalysts in CO-CO2-H2-H2O System[J].Catal Today,1999,51(1):25-38.
[54]范淑蓉,謝筱帆,竇伯生,等.水煤氣變換催化劑Co-Mo-K/γ-Al2O3的酸性對活性的影響[J].應用化學,1991,8(3):42-45.
[55]Hou P,Meeker D,Wise H.Kinetic Studies with a Sulfur-Tolerant Water Gas Shift Catalyst[J].J Catal,1983,80(2):280-285.
[56]Nikolova D,Edreva-Kardjieva R,Gouliev G,et al.The State of(K)(Ni)Mo/γ-Al2O3Catalysts After Water-Gas Shift Reaction in the Presence of Sulfur in the Feed:XPS and EPR Study[J].Appl Catal,A,2006,297(1):135-144.
[57]Eijsbouts S,Mayo S W,Fujita K.Unsupported Transition Metal Sulfide Catalysts:From Fundamentals to Industrial Application[J].Appl Catal,A,2007,322(1):58-66.
[58]Frizi N,Blanchard P,Payen E,et al.Genesis of New HDS Catalysts Through a Careful Control of the Sulfidation of Both Co and Mo Atoms:Study of Their Activation Under Gas Phase[J].Catal Today,2008,130(2-4):272-282.
[59]Frizi N,Blanchard P,Payen E,et al.Genesis of New Gas Oil HDS Catalysts:Study of Their Liquid Phase Sulfidation[J].Catal Today,2008,130(1):32-40.
[60]王月霞.加氫催化劑的器外預硫化[J].煉油設計,2000,30(7):57-58.
[61]van Gesetl J,Leglise J,Duehet J C.Catalytic Properties of a CoMo/Al2O3Catalyst Pre-Sulfided with Alkylpoyl Sulfides:Comparison with Conventional Sulfiding[J].J Catal,1994,145(2):429-436.
[62]Lian Yixin,Wang Huifang,Fang Weiping,et al.Water Gas Shift Activity of Co-Mo/MgO-Al2O3Catalysts Presulfided with Ammonium Sulfide[J].J Nat Gas Chem,2010,19(1):61-66.
[63]Yoosuk Boonyawan,Kim Jae Hyung,Song Chunshan,et al.Highly Active MoS,CoMoS and NiMoS Unsupported Catalysts Prepared by Hydrothermal Synthesis for Hydrodesulfurization of 4,6-Dimethyldibenzothiophene[J].Catal Today,2008,130(1):14-23.
[64]柴永明,趙會吉,柳云騏,等.四硫代鉬酸銨制備方法的改進[J]. 無機鹽工業,2007,39(5):12-15.
[65]高善彬,董群,劉濱,等.預硫化加氫催化劑鈍化機理的研究進展[J].化學工業與工程,2008,25(2):183-188.
[66]王宏悅,潘兆德.器外預硫化耐硫變換催化劑的研制[J].煤化工,2009,140(1):18-20.
[67]楊余芳.一氧化碳加壓變換系統的探討與設計[D].湘潭:湘潭大學,2002.
[68]Heidebrecht P,Sundmacher K.Thermodynamic Analysis of a Cyclic Water Gas-Shift Reactor(CWGSR)for Hydrogen Production[J].Chem Eng Sci,2009,64(23):5057-5065.
[69]王明華,李政,倪維斗.煤制甲醇CO變換工藝組合方式的研究[J].化學工程,2008,36(7):66-70.
[70]侯引平.中小型合成氨裝置的變換工藝[D].天津:天津大學,2005.
[71]Chen Wei-Hsin,Lin Mu-Rong,Jiang Tsung Leo,et al.Modeling and Simulation of Hydrogen Generation from High-Temperature and Low-Temperature Water Gas Shift Reactions[J].Int J Hydrogen Energy,2008,33(22):6644-6656.
[72]Levent M.Water-Gas Shift Reaction over Porous Catalyst:Temperature and Reactant Concentration Distribution[J].Int J Hydrogen Energy,2001,26(6):551-558.
Co-Mo-Based Catalyst and Catalytic Reaction Process for Water-Gas Shift
Lian Yixin,Yang Yiquan,Fang Weiping
(College of Chemistry and Chemical Engineering,National Engineering Laboratory for Green Chemical Productions of Alcohols,Ethers and Esters,Xiamen University,Xiamen Fujian 361005,China)
The development background,present situation and developing vista for CO shift catalysts,and the process technologies for water-gas shift reaction both at home and abroad were introduced.The mechanism of the water-gas shift reaction and the characteristic of the catalyst sulfuration methods were also expounded.By comparing the industrial applications of the catalysts in four cross-sectional shift processes,i.e.the high temperature shift,the high-low temperature shift,the high-low-low temperature shift and the total low temperature shift,it can be imagined that to investigate and develop the Co-Mo-based shift catalysts with high activity and stability,which suit the total low-temperature shift process with energy saving,and the preparation procedures for the catalyst pre-sulfuration will be the developing direction from now on.
water-gas shift;cobalt-molybdenum-based catalyst;total low temperature shift;carbon monoxide;hydrogen;carbon dioxide
1000-8144(2011)04-0347-11
TQ 546.4
A
2010-09-19;[修改稿日期]2010-12-25。
連奕新(1971—),男,福建省惠安縣人,博士,工程師,電郵lianyx@xmu.edu.cn。聯系人:方維平,電話0592-2186291,電郵wpfang@xmu.edu.cn。
本文從水煤氣變換催化劑的現狀與發展出發,著重介紹了目前兩類高效耐硫Co-Mo基變換催化劑的應用與研究情況,闡述了Co-Mo基催化劑的催化機理與硫化方法,進而對比了不同變換工藝流程的操作與能耗,指出適合節能型工藝的Co-Mo基耐硫變換催化劑及其預硫化技術是今后的研究方向。
(編輯 王 萍)
猜你喜歡
山東冶金(2019年6期)2020-01-06 07:45:54
世界農藥(2019年2期)2019-07-13 05:55:12
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
銅業工程(2015年4期)2015-12-29 02:48:39
新疆鋼鐵(2015年3期)2015-11-08 01:59:52
應用化工(2014年3期)2014-08-16 13:23:50
