甲醇氧化PtSnCo/C陽極催化劑
2011-12-11 09:30:22李慶武魏子棟陳四國齊學強
物理化學學報 2011年12期
關鍵詞:催化劑
李慶武 魏子棟 陳四國 齊學強 柳 曉 丁 煒 馬 宇
(重慶大學化學化工學院,輸配電裝備及系統安全與新技術國家重點實驗室,重慶400044)
甲醇氧化PtSnCo/C陽極催化劑
李慶武 魏子棟*陳四國 齊學強 柳 曉 丁 煒 馬 宇
(重慶大學化學化工學院,輸配電裝備及系統安全與新技術國家重點實驗室,重慶400044)
通過乙二醇液相分步還原法制備了金屬質量分數為20%的PtSn/C二元及PtSnCo/C三元催化劑.采用X射線衍射(XRD)光譜法、能量散射譜(EDS)對催化劑進行了表征;通過陽極線性伏安掃描法(LSV)、連續循環伏安法(CV)、預吸附單層CO溶出法研究了其電化學性質.結果表明,PtSnCo/C三元催化劑較商業化JM-PtRu/C催化劑具有更好的氧化甲醇催化活性.循環伏安掃描100圈后發現,PtSn/C二元催化劑的甲醇氧化峰電流快速衰減到其初始氧化峰電流的11%左右,而PtSnCo/C三元催化劑僅衰減到其初始值的50%左右,這表明PtSnCo/C三元催化劑具有更好的化學穩定性.在PtSnCo/C催化劑上,甲醇氧化起始電位比直接吸附CO后的CO陽極溶出電位負,意味著甲醇在PtSnCo/C催化劑上氧化的中間產物不是CO,而是比CO更為活潑且易于氧化的中間物種.
直接甲醇燃料電池;PtSnCo/C;PtSn/C;穩定性;甲醇氧化
1 引言
直接甲醇燃料電池(DMFC)因其具有質量輕、體積小、結構簡單、比能量密度高、低溫操作等優點,在移動電源、電動汽車等領域具有廣泛前景.1,2但困擾DMFC商業化發展的三個主要問題是:3(1)甲醇在陽極上氧化反應速率緩慢;(2)甲醇自陽極透過質子交換膜向陰極滲透使電池陰極性能下降;(3)甲醇氧化產物CO2的液封效應問題.發展性能優異的催化劑是克服甲醇氧化反應速率緩慢的最有效手段.在酸性溶液中,甲醇氧化最有效的催化劑是Pt或Pt基合金催化劑.然而,甲醇氧化反應產生的CO中間體強吸附在Pt表面形成Pt-COads而使其失活.一般認為,4,5鉑電極只有在相對參比氫電極(RHE)在0.5 V以上才能形成含氧物種使COads進一步氧化為CO2,從而使其恢復活性.因此,提高Pt對甲醇氧化反應活性的關鍵是在低電位下高效地氧化去除CO以釋放出Pt的活性位.6
目前的解決辦法是在Pt中加入一種或幾種能在低電位下生成含氧物種的金屬形成二元或多元的Pt基合金催化劑.7,8PtRu/C是公認的商業化效果最好的DMFC陽極電催化劑.Ru原子促進CO氧化的雙功能機理已被普遍認可,9-12即在比Pt電位低0.2-0.3 V的電位下,Ru表面能形成含氧物種,從而實現在較低的電位下氧化去除CO中間體.Goodenough等13提出PtRu中Pt電子結構的改變是其抗CO中毒能力改善的另一種解釋.PtSn是僅次于PtRu而倍受關注的另一個二元催化劑.有研究報道在Pt上欠電位沉積Sn原子且Sn的覆蓋率在20%左右時,發現其對Pt在較低電位下發生的甲醇氧化反應表現出了明顯的催化增強作用,認為Sn的作用也同樣可以用雙功能機理和電子效應來解釋.14,15然而,錫對鉑的甲醇氧化催化活性是否具有增強作用還持有爭議.如Frelink等16發現PtSn合金催化劑并沒有表現出比Pt更優異甚至更差的甲醇氧化催化活性,有報道認為這是由于錫的存在不利于Pt上的甲醇吸附以及脫氫過程.17此外,酸性燃料電池操作條件下PtSn合金催化劑中Sn易于溶解脫出,因此, PtSn二元合金催化劑催化甲醇氧化穩定性還有待進一步提高.PtRuSn,18PtRuCo,19PtSnNi20等三元合金催化劑相繼被報道,均發現三元合金催化劑具有更好的甲醇氧化催化活性、抗CO中毒能力以及電化學穩定性.Travitsky等21通過熱酸處理形成Pt74CoSn26近似于核-殼結構的三元催化劑,用于直接甲醇燃料電池正極氧還原催化且有耐透醇能力,即只對氧還原有催化作用,對甲醇氧化無催化作用.然而Beyhan等22使用原位紅外光譜觀察到PtSnCo催化乙醇氧化時不僅較純Pt上吸附了更少的類CO中間體以及乙酸等吸附物,而且在乙醇氧化過程中產生更多的CO2,表明與純Pt相比,PtSnCo對乙醇等小分子有機物具有更高的催化活性.可見,三元催化劑PtSnCo是否具有催化小分子醇氧化的能力因合成方法、原子間比、結構狀態不同而不同.
本文通過乙二醇液相分步還原法制備了金屬質量分數為20%的PtSnCo/C三元催化劑.采用XRD、EDS對催化劑進行了物理化學表征,通過陽極線性伏安掃描法、預吸附單層CO溶出法、連續循環伏安法研究了其電化學性質.特別探討了Co的加入對PtSn/C二元催化劑的電化學性質的影響,其結果對改善PtSn/C二元催化劑的性能有啟示意義.
2 實驗部分
2.1 試劑與儀器
如無特別說明所用試劑均為分析純:硝酸鈷(Co2(NO3)3·9H2O)、氯化亞錫(SnCl2·2H2O)、濃硫酸,濃鹽酸均為重慶無機化學試劑廠產品;硼氫化鈉(NaBH4,天津市化學試劑研究所);檸檬酸三鈉(TCD),甲醇、無水乙醇、甲酸、氫氧化鈉(重慶川東化學試劑廠);氯鉑酸(H2PtCl6·6H2O,上海精細化工材料研究所);氮氣(重慶泰和氣體實業有限責任公司,99.999%);商業化vulcan X72碳粉(美國Cabot Corp.);20% Pt50Ru50/C,40%Pt/C催化劑(美國Johnson-matthey Corp.);0.5%Nafion(美國Dupont Corp);所有的電解質溶液均用超純水(18.2 MΩ·cm)來配制.
碳紙TGP-H-090(日本Torry Corp公司);自壓反應釜RD-50(中國石油化工股份有限公司石油化工科學研究院);電化學工作站AutoLab(荷蘭,Metrohm Singapore Pte Ltd.);XRD-6000型X射線衍射儀(日本島津公司);FEI Nova 400型掃描電子顯微鏡(Peabody,MA,Netherland).
2.2 催化劑的制備
Vulcan XC-72碳粉的預處理按照文獻23進行,催化劑制備具體過程如下:取80 mg預處理后的Vulcan XC-72碳粉,5.2 mg SnCl2·2H2O,20 mg Co(NO3)2·9H2O和一定量的TCD(TCD和金屬摩爾比為2:1)加入到20 mL乙二醇中,超聲攪拌0.5 h使金屬鹽充分溶解于乙二醇中,調節pH~11,然后在強烈攪拌下將5 mL濃度為8 g·L-1(NaBH4/乙二醇)溶液滴加到上述體系中,攪拌反應1 h后使Sn2+和Co2+還原于碳粉上,再向上述溶液中加入0.7 mL濃度為40 g·L-1的H2PtCl6水溶液(保持Pt:Sn:Co的原子比為3:1:3),調節pH~11,分散后轉移入自壓反應釜中, 160°C下高溫反應6 h,取出自然降溫后,抽濾,用1: 1無水乙醇/水混合溶液洗滌數次,除掉氯離子和多余的TCD,60°C真空干燥24 h后待用.所制備的催化劑標記為PtSnCo/C.使用相同的方法及步驟制備PtSn/C催化劑,催化劑中Pt:Sn的理論原子比為3:1.
2.3 催化劑的物理化學性質表征
催化劑晶相結構和晶粒大小的分析是在XRD-6000X射線衍射儀上進行的,衍射源為CuKα靶(λ= 0.1542 nm),測試角度為10°-80°,掃描速率為2(°)· min-1.催化劑上Pt納米顆粒的平均粒徑可以通過Scherrer公式計算.
d=0.9λ/(B·cosθ) (1)上式中λ為入射光波長(0.1542 nm),B是以弧度表示的衍射峰半峰寬(FWHM),θ為衍射峰出現位置的角度.d為引起該衍射晶面的法線方向上的晶面尺寸(nm).對于晶體平均粒徑大小的計算,一般來講,如是較大顆粒的晶體則應該選擇較高晶面指數的半峰寬;相反,如果較小細微晶體,則應該選擇較低晶面指數的半峰寬.同時,盡量選擇受干擾小的晶面特征峰的半峰寬.本文選擇Pt(111)和Pt(200)兩個晶面特征峰的平均值來估算Pt晶粒平均粒徑大小.
EDS表征是在FEI Nova 400掃描電鏡下進行,選取三個區域取其平均值來計算催化劑中Pt、Sn、Co的原子比.
2.4 電催化性能評價
采用線性掃描伏安法(LSV)、循環伏安法(CV)和預吸附單層CO溶出法研究催化劑的電化學性能,并與英國Johnson-Matthey公司的商業Pt/C催化劑(記為JM-Pt/C)進行比較.電化學測試采用三電極體系在AutoLab電化學工作站上進行,其中輔助電極為鉑絲,參比電極為Ag/AgCl(飽和KCl溶液)電極(本文電位值無特別說明均是相對于Ag/AgCl電極電位).電解質分別為0.5 mol·L-1H2SO4溶液以及0.5 mol·L-1H2SO4+0.5 mol·L-1CH3OH.電化學測試中持續向電解質中通入氮氣以驅除溶解氧,所有測試均在室溫下進行.
預吸附單層CO溶出實驗.先向電解質中通高純氮0.5 h除氧,以50 mV·s-1在-0.15-1.05 V范圍內掃描10圈使電極表面達到穩定,再向電解質中通入CO(實驗室甲酸和濃硫酸制備CO)0.5 h,使電極表面上CO的吸附達到飽和,停止通CO,再用高純氮除去溶液中的CO,然后進行循環伏安掃描,掃描速率為50 mV·s-1.
工作電極制備:稱取2 mg催化劑,加入400 μL無水乙醇和適量0.5%Nafion,超聲波振蕩30-60 min,混合至成均勻墨水狀液體,取100 μL懸浮液涂于按照文獻24處理的TGP-H-090碳紙上(1 cm×1.5 cm),涂平,80°C烘干,即得到工作電極.相應催化劑制備的電極,記為相應的電極,如PtSn/C催化劑制備的電極記為PtSn/C電極.
3 結果與討論
3.1 催化劑的XRD及EDS表征
圖1是各催化劑的XRD圖.從圖1可以看出在2θ為40.4°、44.5°、68.2°處分別對應Pt的(111)、(200)、(220)晶面衍射峰.與標準譜圖(JCPDS-652868)對比發現,PtSn/C和PtSnCo/C催化劑均為Pt的面心立方晶系.在衍射圖譜中沒有出現Sn、SnO2、Co、CoO2、Co3O4等特征衍射峰,推測Sn或Co在催化劑中是以無定形態或固溶體形式存在.根據XRD估算的JM-Pt/C,PtSn/C,PtSnCo/C的平均粒徑分別為3.1、3.2、2.3 nm.
對上述催化劑的EDS分析發現,PtSn/C催化劑中Pt:Sn原子比為3.53:1,基本接近制備時的配料比.但PtSnCo/C催化劑Pt:Sn:Co原子比為4.06:1.26: 1(見圖2),Pt:Sn原子比基本接近于制備時的配料比,Co的含量遠低于理論值.我們推測這是因為Co2+是三種金屬離子里是最難被還原的,還原速率的不一致導致了Co偏離配料值.此外,檸檬酸三鈉的絡合作用進一步導致Co2+難以還原.

圖2 PtSnCo/C催化劑的EDS分析Fig.2 EDS spectra of PtSnCo/C Catalyst
3.2 催化劑的電化學測試
圖3是各電極在0.5 mol·L-1H2SO4+0.5 mol· L-1CH3OH溶液陽極線性掃描伏安曲線.從圖3看出PtSn/C電極的甲醇氧化起始電位與商業化的JM-Pt/ C電極基本一致,并沒有明顯的提升甲醇氧化反應的活性,表明Sn的加入并沒有增強Pt的甲醇氧化催化活性,這與Frelink等16的觀察現象是一致的.然而,當形成PtSnCo/C三元催化劑后,其甲醇氧化起始電位比PtSn/C電極明顯負移了約150 mV,與商業化JM-PtRu/C電極的甲醇氧化起始電位一致,但其峰電流密度是所有電極中最高的,說明Co的加入有效提升了PtSnCo/C電極的甲醇氧化催化活性.

圖3 各電極在0.5 mol·L-1H2SO4+0.5 mol·L-1CH3OH電解液中的線性伏安曲線Fig.3 Anodic linear sweep voltammograms of the methanol electrooxidation reaction on different electrodes in 0.5 mol·L-1H2SO4+0.5 mol·L-1CH3OH electrolytescan rate:5 mV·s-1

圖4 在0.5 mol·L-1H2SO4溶液中不同電極上預吸附單層CO的溶出伏安曲線Fig.4 Anodic stripping voltammograms of pre-adsorbed CO monolayer on differenct electrodes in 0.5 mol·L-1 H2SO4solutionscan rate:50 mV·s-1.1st cycle(black solid line)for the electrooxidation of CO,2nd cycle(red dot line)after removal of CO
通常認為,甲醇氧化過程的主要中間體是CO.圖4是各電極上預吸附CO后的陽極溶出實驗.從圖4中可以看出JM-Pt/C上CO氧化起始電位在0.62 V左右,與文獻報道值0.7 V(vsRHE)25接近.PtSn/C的CO氧化起始電位與JM-Pt/C相比負移了約100 mV (0.5 V左右),表明其比鉑更容易在低電位下催化氧化CO.這是因為Sn能在比Pt更低的電位下形成Sn-OHads物種,17從而降低CO的電位.但與商業化JM-PtRu/C(0.3 V左右)相比,PtSn/C的氧化CO的電位明顯偏高.而PtSnCo/C的CO氧化起始電位與PtSn/C相比略負,但不及商業化JM-PtRu/C的CO氧化起始電位負.說明Co的加入并不能進一步降低PtSn/C催化劑的CO氧化電位.圖3和圖4的結果顯示,在JM-PtRu/C電極上,甲醇和CO的起始氧化電位基本一致,大約0.3 V.而在Pt/C、PtSn/C、PtCo/C、PtSnCo/C電極上,甲醇氧化起始電位均比直接吸附CO后的CO陽極溶出電位負,很可能是甲醇在這些電極上氧化的中間產物是比CO更為活潑且易于氧化的中間物種,如CHO(COH)、CH3O、HCO、CH2O,26-28或者甲醇氧化產生的吸附態CO并不完全等同于從CO氣體吸附到催化劑表面的CO.類似的現象在孫世剛等29的研究中也有觀察.Co加入后, PtSnCo/C電極相對于Pt/C、PtSn/C、PtCo/C電極甲醇氧化電流顯著增加,是晶粒細化,電極表面活性位增加所致(見圖1 XRD表征的晶粒尺度).
圖5是PtSn/C和PtSnCo/C電極分別在0.5 mol· L-1H2SO4+0.5 mol·L-1CH3OH溶液中的連續循環伏安曲線,記錄了100圈的實驗數據(如圖5(a,b)).通過對連續循環伏安掃描過程中催化劑的甲醇氧化正向峰電流與反向峰電流的比值If/Ib分析發現(圖5c),PtSnCo/C和PtSn/C的If/Ib的初始值基本相同,但PtSnCo/C三元催化電極的If/Ib的值隨掃描圈數的增加而基本不變,進一步說明其具有更好的電化學穩定性.30-32雖然PtSn/C電極的If/Ib值隨掃描圈數增加而逐漸增大,似乎意味著表面毒性中間體氧化電流相對甲醇正向氧化電流越來越少.但是,圖5(d)顯示,經過100圈連續循環伏安掃描后,PtSn/C電極的甲醇氧化峰電流If衰減到初始氧化峰電流If0的11%左右,而PtSnCo/C電極僅衰減到初始值的50%左右,說明PtSn/C電極的活性位(面積)顯著下降, PtSnCo/C電極卻具有更好的化學穩定性.
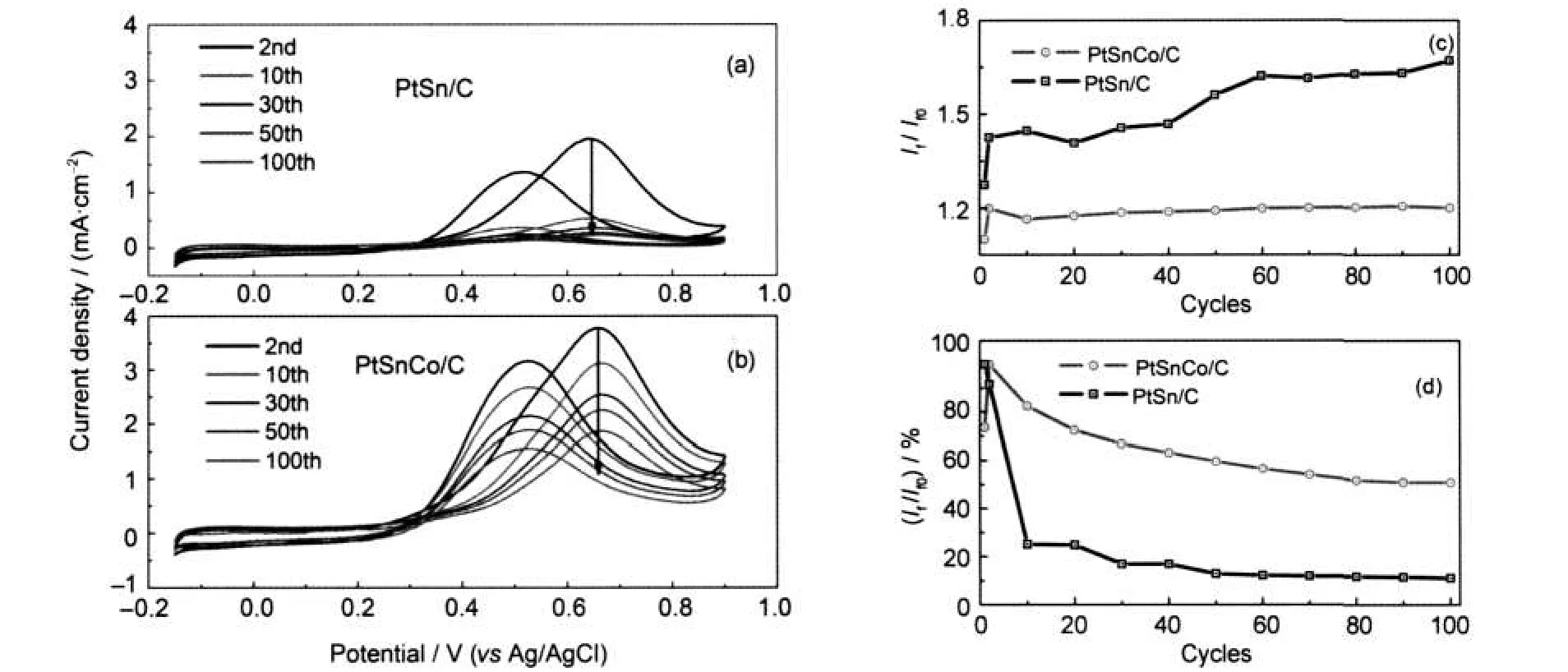
圖5 PtSn/C和PtSnCo/C電極在0.5 mol·L-1H2SO4+0.5 mol·L-1CH3OH溶液中的連續循環伏安曲線圖(a,b)以及峰電流If/Ib(c)和If/If0(d)隨掃描圈數的變化Fig.5 Cyclic voltammograms(a,b)of electrodes PtSn/C and PtSnCo/C recorded in 0.5 mol·L-1H2SO4+0.5 mol·L-1CH3OH electrolyte with N2bubbled at scan rate:10 mV·s-1,ratio of If/Ib(c)and If/If0(d)with cycle numbers on two electrodesIf:the forward peak current,Ib:the backward peak current,If0:the initial current of the forward current
4 結論
探討了Co的加入對PtSn/C二元催化電極的電化學性質的影響.結果表明,(1)PtSnCo/C三元催化劑具有與商業化PtRu/C催化劑相近的甲醇氧化催化活性,與PtSn/C二元催化劑相比具有更高的催化甲醇氧化活性和穩定性.Co加入后,PtSnCo/C催化劑相對于Pt/C、PtSn/C、PtCo/C催化劑,甲醇氧化電流顯著增加,是晶粒細化,電極表面活性位增加的結果.(2)在PtSnCo/C催化劑上,甲醇氧化起始電位比直接吸附CO后的CO陽極溶出電位負,意味著甲醇在PtSnCo/C催化劑上氧化的中間產物是比CO更為活潑且易于氧化的中間物種,或者不完全等同于從CO氣體吸附到催化劑表面的類CO物種.考慮到錫、鈷比釕價廉,三元催化劑PtSnCo/C作為甲醇氧化陽極催化劑前景更好.
(1) Prater,K.B.J.Power Sources 1996,61,105.
(2)Doo,H.J.;Chang,H.L.;Chang,S.K.;Dong,R.S.J.Power Sources 1998,71,169.
(3) Xu,Q.J.;Zhou,X.J.;Li,Q.X.;Li,J.G.Acta Phys.-Chim.Sin. 2010,26,2135.[徐群杰,周小金,李巧霞,李金光.物理化學學報,2010,26,2135.]
(4) Beden,B.;Lamy,C.;Bewick,A.;Kunimatsu,K.J.Electroanal. Chem.1981,121,343.
(5) Hamnett,A.Catal.Today 1997,38,445.
(6) Li,L.L.;Wei,Z.D.;Yan,C.;Luo,Y.H.;Yin,G.Z.;Sun,C.X. Acta Phys.-Chim.Sin.2007,23(5),723.[李蘭蘭,魏子棟,嚴 燦,羅義輝,尹光志,孫才新.物理化學學報,2007,23(5), 723.]
(7)An,X.S.;Chen,D.J.;Zhou,Z.Y.;Wang,Q.;Fan,Y.J.;Sun,S. G.Acta Phys.-Chim.Sin.2010,26,1207.[安筱莎,陳德俊,周志有,汪 強,樊友軍,孫世剛.物理化學學報.2010,26, 1207.]
(8)Wei,Z.D.;Guo,H.T.;Tang,Z.Y.J.Power Sources 1996,58, 239.
(9)Chrzanowski,W.;Kim,H.;Wieckowski,A.Catal.Lett.1998, 50,69.
(10) Wei,Z.D.;Li,L.L.;Luo,Y.H.;Yan,C.;Sun,C.X.;Yin,G.Z.; Shen,P.K.J.Phys.Chem.B 2006,110,26055.
(11) Chrzanowski,W.;Wieckowski,A.Langmuir 1997,13,5974.
(12) Zhou,W.J.;Zhou,Z.H.;Li,W.Z.;Sun,G.Q.;Xin,Q. Chemistry 2003,66(4),228. [周衛江,周振華,李文震,孫公權,辛 勤.化學通報.2003,66(4),228.]
(13)Goodenough,J.B.;Manoharan,R.;Shukla,A.K.;Ramesh,K. V.Chem.Mater.1989,1,391.
(14)Wei,Z.D.;Miki,A.;Ohmori,T.;Osawa,M.Acta Phys.-Chim. Sin.2002,18(12),1120.[魏子棟,三木敦史,大森唯義,大澤雅致.物理化學學報,2002,18(12),1120.]
(15) She,C.X.;Li,X.Q.;Ren,B.;Lin,H.S.;Tian,Z.Q.Chinese Journal of Light Scattering 2002,3,223.[佘春興,李筏琴,任 斌,林華水,田中群.光散射學報,1999,3,223]
(16) Frelink,T.;Visschefz,W.;Van Veen,J.A.R.Electrochim.Acta 1994,39,1871.
(17) Antolini,E.;Gonzalez,E.R.E.Catal.Today 2011,160,28.
(18)Neto,A.O.;Dias,R.R.;Tusi,M.M.;Lindardi,M.;Spinacé,E. V.J.Power Sources 2007,166,87.
(19) Strasser,P.J.Comb.Chem.2008,10,216.
(20) Spinacé,E.V.;Lindardi,M.;Neto,A.O.Electrochem. Commun.2005,7,365.
(21) Travitsky,N.;Burstein,L.;Rosenberg,Y.;Peled,E.J.Power Sources 2009,194,161.
(22) Beyhan,S.;Kadirgan,F.;Léger,J.M.In-situ Infrared Spectroscopy Study of Ethanol Oxidation on Pt and PtSn-Based TrimetallicAnode Electrocatalysts for Direct Ethanol Fuel Cell. In Electrode Processes Relevant to Fuel Cell Technology.217th ECS Meeting,Vancouver,Canada,April 25-30,2010;Birss,V.; Kulesza,P.;Mustain,W.;Ota,K.;Wilkinson,D.; The Electrochemical Society 2010,B7,603.
(23) Wei,Z.D.;Chen,S.G.;Liu,Y.;Sun,C.X.;Shao,Z.G.;Shen, P.K.J.Phys.Chem.C 2007,111,15456.
(24) Liao,M.J.;Wei,Z.D.;Chen,S.G.;Li,L.;Ji,M.B.;Wang,Y. Q.Int.J.Hydrog.Energy 2010,35,8071.
(25) Crabb,E.M.;Marshall,R.;Thompsett,D.J.Electrochem.Soc. 2000,147,4440.
(26) Xia,X.H.;Iwasita,T.;Ge,F.;Vielstich,W.Electrochim.Acta 1996,41,711.
(27) Iwasita,T.;Braz.J.Chem.Soc.2002,13,401.
(28) Wang,J.;Masel,R.I.Surf.Sci.1991,235,199.
(29)Zhou,Z.Y.;Tian,N.;Zeng,D.M.;Sun,S.G.The Proceeding of 12th National Conference of Electrochemistry,Shanghai, China,2003,A040.[周志有,田 娜,曾冬梅,孫世剛.第12次全國電化學會議論文集,上海,2003,A040.]
(30) Chen,W.;Kim,J.;Sue,S.;Chen,S.Langmuir 2007,23,11303.
(31) Chen,J.;Wang,M.;Liu,B.;Fan,Z.;Cui,K.;Kuang,Y.J.Phys. Chem.B 2006,110,1775.
(32) Hsieh,C.T.;Lin,J.Y.J.Power Sources 2009,188,347.
May 4,2011;Revised:August 25,2011;Published on Web:September 5,2011.
PtSnCo/C Anode Catalyst for Methanol Oxidation
LI Qing-Wu WEI Zi-Dong*CHEN Si-Guo QI Xue-Qiang LIU Xiao DING Wei MAYu
(State Key Laboratory of Power Transmission Equipment&System Security and New Technology,School of Chemistry and Chemical Engineering,Chongqing University,Chongqing 400044,P.R.China)
Abinary metallic catalyst(PtSn/C)and a ternary metallic catalyst(PtSnCo/C)with a metal mass fraction of 20%were prepared by borohydride reduction and subsequent hydrothermal treatment in a glycol liquid phase.The structure and composition of the as-prepared electrocatalysts were characterized by X-ray diffraction(XRD)and energy-dispersive spectrometry(EDS).Their activity and stability for the catalysis of methanol oxidation were evaluated by anodic linear sweep voltammetry(LSV),cyclic voltammetry(CV),and the anodic stripping of a pre-adsorbed CO monolayer.We found that the PtSnCo/C catalyst gave the best catalytic activity for the methanol oxidation of all the catalysts including the commercial JM-PtRu/C catalyst. After 100 cycles,the peak current of methanol oxidation for the PtSn/C catalyst rapidly decreased to 11%of its initial peak current but PtSnCo/C decreased to only 50%.This result suggests that the PtSnCo/C catalyst has better chemical stability for the catalysis of methanol oxidation compared to the PtSn/C catalyst.The more negative onset potential of methanol oxidation for the PtSnCo/C catalyst relative to pre-adsorbed CO oxidation implies that the intermediates of methanol oxidation on the PtSnCo/C catalyst may be ones,which can bemoreeasily oxidized thanCO,instead ofCO.
Dierct methanol fuel cell;PtSnCo/C;PtSn/C;Stability;Methanol oxidation
10.3866/PKU.WHXB20112857
?Corresponding author.Email:zdwei@cqu.edu.cn;Tel:+86-23-65105160.
The project was supported by the National Natural Science Foundation of China(20906107,20936008),Innovative Talent Training Project, Chongqing University,China(101061136),and Fundamental Research Funds for the Central University,China(CDJXS10221141,11132229).
國家自然科學基金(20906107,20936008),重慶大學985創新人才培養建設計劃(101061136)和中央高校基礎研究基金(CDJXS10221141, 11132229)資助項目
O643.36
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
