具有π共軛骨架的Salen-卟啉型同、異雙核配合物的合成及譜學性質
2012-03-06 04:43:14王官耀閆偉偉張曉紅阮文娟朱志昂
物理化學學報 2012年12期
王官耀 閆偉偉 張曉紅 阮文娟 朱志昂
(南開大學化學學院,天津300071)
1 引言
配位化學是目前最活躍的學科之一,近年來,隨著配位化學研究的深入,具有多樣化結構的多核金屬配合物的研究日趨受到重視.1-5以金屬配合物為基礎的各種功能材料發展十分迅速,對多核金屬配合物性質的研究可以為開發制備新型的光、電、磁等功能材料提供實驗和理論指導.卟啉及其金屬配合物作為一類特殊的大環共軛體系,具有優越的物理、化學及光學特征,在光電材料、分子識別、分子組裝、有機合成、光譜分析等領域有廣闊的應用前景.6-12而包含另一類常見的配體Salen(N, N?-bis-(saliylaldehyde)ethylendiamine)或其衍生物的配位化合物則主要應用于不對稱催化領域,13-16對Salen型配合物的光、電、磁等各項功能性質的探討近些年來也逐漸成為熱點.17-21既然卟啉類化合物與Salen型配合物均能與一系列的過渡金屬結合形成穩定的化合物,由此可以構建具有特殊光、電、磁等性質的Salen-卟啉型雙核金屬配合物,不僅對開發新型功能材料具有重要意義,而且在光學器件、分子開關、電極修飾及磁性材料等領域也具有重要的潛在應用價值.1,22,23
本文合成了文獻中未見報道的多種具有π共軛骨架的新型Salen-卟啉型金屬配合物,采用1H NMR、紫外-可見吸收光譜、紅外光譜和熒光光譜等多種表征手段對它們進行了研究.
2 實驗部分
2.1 儀器和試劑
Mercury Vx 300 MHz核磁共振儀(美國Varian公司生產),氘代三氯甲烷(CDCl3)為溶劑,四甲基硅烷(TMS)為內標;MAGNA-560傅里葉變換紅外光譜儀(美國Nicolet公司生產);TRACE DSQ型質譜儀(美國Thermofinnigan公司生產);Shimadzu UV-2450紫外-可見分光光度(日本Shimadzu公司生產); Shimadzu RF-5301 PC型熒光光譜儀(日本Shimadzu公司生產).
吡咯、苯甲醛、三乙胺、三氯甲烷均為分析純試劑,使用前按試劑手冊24方法處理.碳酸鉀(分析純)于馬弗爐中400°C焙燒2 h待用,其余試劑也均為分析純,可直接使用.
2.2 合 成
合成路線見示意圖1、2和3.
5,10,15,20-四苯基-6?,7?-二氨基喹啉[2,3-b?]卟啉(化合物A)按照文獻25-28合成,化合物1-8按如下方法合成.
2.2.1 化合物1和2的合成
將3,5-二叔丁基-2-羥基苯甲醛250 mg(1 mmol)溶于10 mL無水甲醇中,加入適量無水碳酸鉀,攪拌下滴加化合物A(80 mg)的三氯甲烷溶液.混合溶液在室溫下避光攪拌3天.過濾,蒸除溶劑,以二氯甲烷/石油醚為1:3(體積比,下同)的混合液作為淋洗劑,用硅膠柱分離提純,收集第一色帶,產率為10%;再以二氯甲烷/石油醚為2:1的混合液為淋洗劑,收集第二色帶,產率為64%.其中第一色帶為化合物2,第二色帶為化合物1.
化合物 1(C65H54N8O):1H NMR(CDCl3,300 MHz),δ-2.56(s,2H,pyrrole N―H),1.41(s,18H, t-Bu―H),4.54(s,2H,―NH2),7.02(s,1H,quinoxaline H),7.35(s,1H,quinoxaline H),7.38(d,J=2.326 Hz,1H,ArO―H),7.57(d,J=2.146 Hz,1H,ArO―H), 7.76-7.79(m,12H,aryl H),8.17(d,J=1.557 Hz,4H, aryl H),8.21-8.24(m,4H,aryl H),8.71(s,2H,β-pyrrolic H),8.77(s,1H,N=CH),8.92-8.93(m,4H, β-pyrrolic H),13.03(s,1H,O―H).MS,m/z 963.64 (M+H)+.
化合物2(C80H74N8O2):1H NMR(CDCl3,300 MHz),δ-2.56(s,2H,pyrrole N―H),1.40(s,18H, t-Bu―H),1.50(s,18H,t-Bu―H),7.35(d,J=7.8 Hz, 2H,ArO―H),7.52(d,J=2.403 Hz,2H,ArO―H), 7.55(s,2H,quinoxaline H),7.79-7.84(m,12H,aryl H),8.19(d,J=7.00 Hz,4H,aryl H),8.22-8.25(m,4H, aryl H),8.72(s,2H,β-pyrrolic H),8.77(s,2H,N=CH),8.96(m,4H,β-pyrrolic H).13.36(s,2H,O―H). MS,m/z 1179.81(M+H)+.
2.2.2 模板法合成化合物3-6
(1)將20 mg化合物1溶于4 mL二氯甲烷中,加入醋酸鎳的甲醇飽和溶液8 mL,避光條件下回流20 min,加入36 mg 3,5-二叔丁基-2-羥基苯甲醛,30 min后有深紫紅色沉淀出現,繼續反應直至沉淀的量不再變化.過濾,將沉淀風干后,用適量二氯甲烷溶解,并用蒸餾水洗滌至水相呈中性.用無水硫酸鎂干燥,蒸除溶劑.用硅膠柱分離提純,以二氯甲烷/石油醚為1:2的混合液為淋洗劑,收集第一色帶,產率為30%;收集第二色帶,產率為45%.二者均為紫紅色固體,其中第一色帶為化合物4,第二色帶為化合物3.

示意圖1 化合物1和2的合成路線Scheme 1 Synthesis route of compounds 1 and 2
化合物3(C80H72N8NiO2):1H NMR(CDCl3,300 MHz),δ-2.59(s,2H,pyrrole N―H),1.42(s,18H, t-Bu―H),1.49(s,18H,t-Bu―H),7.48(d,J=2.371, 2H,quinoxaline H),7.52(s,2H,ArO―H),7.55(s,2H, ArO―H),7.77-7.79(m,4H,aryl H),7.86(s,2H,aryl H),7.88(s,2H,aryl H),7.96-7.98(m,4H,aryl H), 8.16-8.21(m,4H,aryl H),8.24(m,4H,aryl H),8.40 (s,2H,N=CH),8.70(s,2H,β-pyrrolic H),8.93(s, 4H,β-pyrrolic H).MS,m/z 1235.85(M+H)+.

示意圖2 化合物3-6的合成路線Scheme 2 Synthesis route of compounds 3-6

示意圖3 化合物7和8的合成路線Scheme 3 Synthesis route of compounds 7 and 8
化合物4(C80H70N8Ni2O2):1H NMR(CDCl3,300 MHz),δ 1.39(s,18H,t-Bu―H),1.48(s,18H,t-Bu―H),7.18(s,2H,quinoxaline H),7.35(d,J=7.8 Hz,2H, ArO―H),7.47(d,J=2.450 Hz,2H,ArO―H),7.92(s, 4H,aryl H),7.75-7.78(m,4H,aryl H),7.89-7.90(m, 8H,aryl H),7.98(m,4H,aryl H),8.34(s,2H,N=CH),8.63(s,2H,β-pyrrolic H),8.69(s,4H,β-pyrrolic H).MS,m/z 1291.64(M+H)+.
(2)將20 mg化合物1溶于4 mL二氯甲烷中,加入醋酸鎳的甲醇飽和溶液8 mL,避光條件下回流20 min,加入20 μL水楊醛,45 min后有深紫紅色沉淀出現,繼續反應直至沉淀的量不再變化為止.過濾,將沉淀風干后,用適量二氯甲烷使之溶解,蒸餾水洗滌至水相呈中性.用無水硫酸鎂干燥,蒸除溶劑.用硅膠柱分離提純,以二氯甲烷/石油醚為1:2的混合液為淋洗劑,收集第一色帶,產率為69%;再以二氯甲烷/石油醚為2:1的混合液為淋洗劑,收集第二色帶,產率為29%,蒸除溶劑得紫紅色固體.二者均為紫紅色固體,第一色帶為化合物6,第二色帶為化合物5.
化合物5(C72H56N8NiO2):1H NMR(CDCl3,300 MHz),δ-3.44(s,2H,pyrrole N―H),1.46(m,9H, t-Bu―H),1.52(m,9H,t-Bu―H),6.73(s,1H,quinoxaline H),6.99(s,1H,quinoxaline H),7.04(s,1H,ArO―H),7.42(s,2H,ArO―H),7.60(s,2H,ArO―H),7.66 (s,1H,ArO― H),7.72-7.77(m,4H,aryl H), 7.80-7.85(m,6H,aryl H),7.88(m,4H,aryl H),7.99 (s,6H,aryl H),8.35(s,1H,N=CH),8.47(s,1H,N=CH),8.57(s,2H,β-pyrrolic H),8.75-8.88(m,4H, β-pyrrolic H).MS,m/z 1123.67(M+H)+.
化合物6(C72H54N8Ni2O2):1H NMR(CDCl3,300 MHz)δ 1.40(s,9H,t-Bu-H),1.48(s,9H,t-Bu―H), 7.04(s,1H,quinoxaline H),7.15(s,1H,quinoxaline H),7.32(s,1H,ArO―H),7.39(s,1H,ArO―H),7.42 (s,1H,ArO―H),7.45(s,1H,ArO―H),7.52(s,1H, ArO―H),7.55(s,1H,ArO―H),7.73-7.76(m,10H, aryl H),7.84-7.86(m,6H,aryl H),7.92-7.94(m,4H, aryl H),8.15(s,1H,N=CH),8.23(s,1H,N=CH), 8.54(s,2H,β-pyrrolic H),8.64-8.69(m,4H,β-pyrrolic H).MS,m/z 1179.302(M+H)+.
2.2.3 化合物7和8的合成
將10 mg化合物3(或5)溶于15 mL二氯甲烷,加入醋酸鋅的甲醇飽和溶液4 mL,避光室溫攪拌,直至TLC顯示反應完成.反應混合物用蒸餾水洗滌三次后,用無水硫酸鎂干燥,蒸除溶劑.以二氯甲烷/石油醚為2:1的混合液為淋洗劑,用硅膠柱分離提純,收集主色帶,得化合物7(或8).
化合物7(C80H70N8NiZnO2):1H NMR(CDCl3,300 MHz)δ 1.44(s,18H,t-Bu―H),1.50(s,18H,t-Bu―H),7.24(s,2H,quinoxaline H),7.33(d,J=4.446 Hz, 2H,ArO―H),7.49(d,J=2.122 Hz,2H,ArO―H), 7.79-7.83(m,4H,aryl H),7.93-7.94(m,4H,aryl H), 8.01-8.02(m,8H,aryl H),8.16-8.17(m,4H,aryl H), 8.34(s,2H,N=CH),8.62(s,2H,β-pyrrolic H), 8.83-8.89(m,4H,β-pyrrolic H).MS,m/z 1296.40(M+ H)+.
化合物8(C72H54N8NiZnO2):1H NMR(CDCl3,300 MHz)δ 1.49(m,9H,t-Bu―H),1.54(m,9H,t-Bu―H),6.51(s,1H,quinoxaline H),6.75(s,1H,quinoxaline H),6.84(s,1H,ArO―H),7.22(s,2H,ArO―H), 7.53(s,2H,ArO― H),7.61(s,1H,ArO― H), 7.75-7.79(m,4H,aryl H),7.87-7.91(m,6H,aryl H), 7.93-8.14(m,10H,aryl H),8.31(s,1H,N=CH),8.42 (s,1H,N=CH),8.50(s,2H,β-pyrrolic H),8.64-8.76 (m,4H,β-pyrrolic H).MS,m/z 1185.31(M+H)+.
3 結果與討論
3.1 核磁共振氫譜
在化合物1中,卟啉環內的吡咯氫的化學位移在-2.56處,側鏈―NH2、CH=N和―OH的質子峰化學位移分別在4.54、8.77和13.03處.在化合物A中,―NH2的質子峰化學位移處于3.96處.可知由化合物A生成化合物1,―NH2的質子峰向低場移動,這是因為化合物1中―NH2與一側的―OH之間形成了氫鍵,H質子受到了很大的去屏蔽效應.29這也說明化合物A中的一個―NH2與醛基發生反應,生成化合物1,質譜結果進一步驗證了這個結論.
在化合物2中,卟啉環內的吡咯氫的化學位移在-2.56處,側鏈中CH=N和―OH的質子峰化學位移分別在8.77和13.36處,并且氫質子的個數都是兩個,沒有出現化合物A中3.96處附近的―NH2的質子峰,在化合物1中出現的4.54處的―NH2的質子峰也同樣消失,說明化合物A中2個―NH2與2個醛基發生反應,生成化合物2,質譜結果同樣驗證了這個結論.
在自由卟啉化合物中,由于卟啉環流效應在環內產生一個對抗外加磁場的感應磁場,受其影響,卟啉環內的H原子處于卟啉環共軛結構的屏蔽區,使其質子的化學位移移向高場,化學位移值小于零.以金屬Ni2+離子為模板,分別與化合物1中存在的胺基及兩種水楊醛衍生物發生配位反應,生成兩類不同的配合物.
在第一類產物3和5中,卟啉環內的H質子峰依然存在,化學位移分別在-2.59和-3.44處;而―OH質子峰則消失.這說明金屬Ni2+離子只與化合物1和相應的水楊醛反應所形成的Salen型席夫堿配體配位,并未嵌入卟啉環內的空穴中,沒有形成金屬卟啉,即生成單核Salen-卟啉型鎳金屬配合物.相應質譜圖中,(M+H)+正離子峰分別為1235.85、1123.67,與其理論值相符合.在第二類產物4和6中,卟啉環內的H質子峰消失,說明了金屬卟啉配合物的形成;而―OH質子峰的消失,同樣說明Salen型金屬配合物的形成.以上結論證實雙核Salen-卟啉型鎳配合物的生成,所對應的質譜(M+H)+正離子峰分別為1291.64和1179.30,與其理論值相符合.
在化合物3和5與金屬Zn2+離子發生配位生成的產物中,原分別位于-2.59處和-3.44處的卟啉環中的H質子峰消失,說明金屬Zn2+與卟啉環配位,即形成了對稱型異雙核Salen-卟啉型配合物7和不對稱型異雙核Salen-卟啉型配合物8,質譜結果進一步驗證了該結論.
對比金屬配位前后的核磁譜圖不難發現,受中心金屬離子親核作用的影響,金屬配合物周圍電子云密度降低,磁屏蔽作用減小,質子峰的化學位移均有向高場移動的趨勢,并且在雙核金屬配合物(化合物4、6和7)中此現象更為明顯.化合物3是化合物2的單核金屬配合物,而化合物4和7則是化合物2的雙核金屬配合物.不難發現,在化合物2中, CH=N的質子峰的化學位移在8.77處,而在化合物3、4和7中,則分別移到了8.40、8.34和8.34處.化合物6相當于金屬Ni2+離子與化合物5的卟啉環配位而成.在化合物5中,CH=N的質子峰的化學位移分別處于8.35和8.47;在化合物6中則分別移到了8.15和8.23.由此可知,卟啉環中心配位的金屬離子對電子云密度的影響不局限于卟啉環,由于整個分子具有π共軛骨架,其對處于卟啉環一側的Salen型配體也產生了較大的影響.
3.2 紅外光譜
紅外光譜采用石蠟糊法,在4500-400 cm-1范圍內攝譜,主要紅外吸收譜帶的歸屬列于表1.
化合物1在3347、965、731 cm-1處出現N―H伸縮振動吸收峰,而在化合物4和6(通過模板法得到)的紅外光譜中,這三條特征譜帶消失,同時在1009和1007 cm-1處分別出現一個新的強伸縮振動峰,該峰歸屬于Ni―N鍵引起的卟啉環的振動,30這說明卟啉環上的N―H兩個質子被金屬Ni2+離子取代而形成穩定的金屬卟啉配合物.另外化合物4和6分別在1618和1613 cm-1出現的強C=N伸縮振動峰,對比化合物1處于1610 cm-1處的強C=N伸縮振動峰,可知金屬配合物的振動吸收峰向低波數方向移動,說明金屬Ni2+離子與偶氮甲烷基的N原子之間形成了Ni―N鍵.31伴隨―OH伸縮振動峰的消失,在550和420 cm-1左右出現的兩個峰分別歸屬于Ni―O鍵和Ni―N鍵的特征振動峰.這說明了化合物4和6中含有席夫堿結構,證明了化合物4和6是雙核鎳Salen-卟啉型金屬配合物的結論.
化合物3和5的N―H伸縮振動特征吸收峰的三條譜帶依舊存在,這說明兩者的卟啉環內均無金屬離子與吡咯氮原子配位.在1615 cm-1左右均出現強C=N伸縮振動峰,而―OH伸縮振動峰消失的同時,出現550和420 cm-1左右的Ni―O鍵和Ni―N鍵的特征振動峰,則說明化合物3和5存在席夫堿結構,證明了化合物3和5是單核Salen-卟啉型鎳金屬配合物的結論.
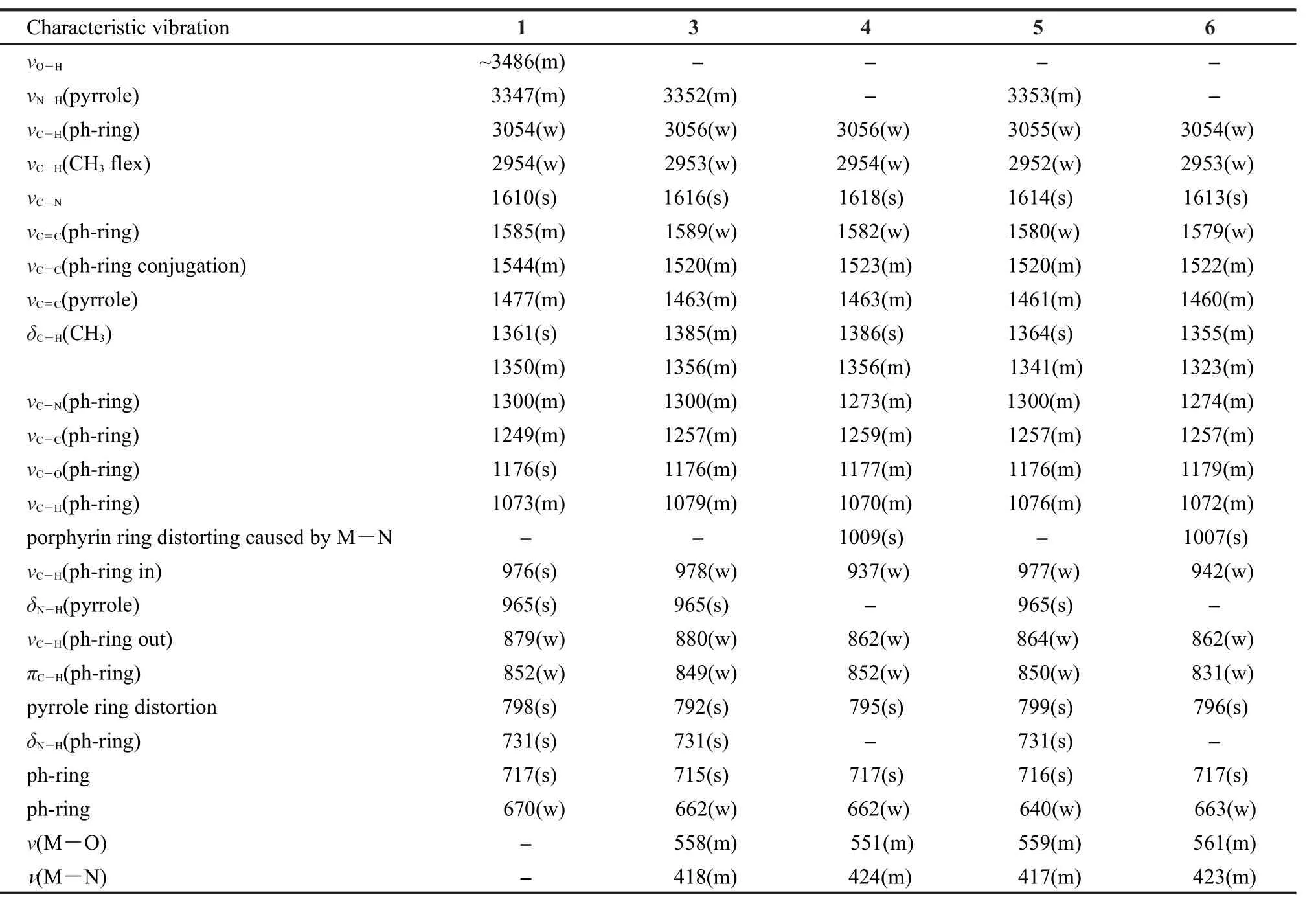
表1 化合物1、3、4、5、6的FTIR特征振動值(單位:cm-1)Table 1 FTIR characteristic vibration values(in cm-1)of compounds 1,3,4,5,6
3.3 紫外-可見光譜
化合物1-8的紫外-可見光譜數據列于表2.
卟啉類化合物在可見光區的電子吸收是由卟啉環大π鍵共軛體系的π-π*躍遷所產生的.卟啉一般具有特殊的紫外-可見吸收,自由卟啉具有一個較強的Soret帶和四個Q帶.當卟啉環內吡咯氮原子與金屬離子配位后,Soret帶將會發生移動,Q帶也會發生變化,最顯著的就是Q帶數目的減少.32
化合物2的吸收光譜在440 nm附近出現的強吸收帶為Soret帶,由卟啉部分a1u(π)-eg(π*)的躍遷而產生.在528、564、600、651 nm處出現的四個較弱吸收帶為Q帶,由卟啉的a2u(π)-eg(π*)的躍遷而產生.33,34卟啉部分與金屬離子配位形成金屬配合物后,卟啉環的對稱性增強,Q帶數目減少.對比化合物2的Soret帶與Q帶,金屬離子取代卟啉環內的兩個氫原子后,Soret帶發生了藍移,Q帶數目減少為兩個.35
Salen型化合物的紫外-可見光譜的特征峰一般可歸為這樣幾類,處于250-280 nm區域的吸收峰為苯環的π-π*躍遷所產生;在300-320 nm區域的吸收峰可歸屬于偶氮甲烷基生色團的π-π*躍遷所引起;而金屬配合物一般在400-500 nm的吸收譜帶則是由于金屬與Salen型配體的配位原子N2O2間的d-π*電荷轉移躍遷所產生;600 nm以上部分有時可觀察到d-d躍遷所致的弱電子吸收峰.Salen型金屬配合物的電子吸收譜帶發生紅移或藍移與金屬離子的電負性及金屬本身內在的性質有關.根據現代分子軌道理論,很多因素都可以影響整個共軛體系的電子云密度,從而使吸收峰發生不同程度的紅移或藍移.36-42
本文設計合成的Salen-卟啉型化合物,由于其具有π共軛骨架,卟啉和Salen的特征峰與卟啉和Salen單體化合物相比會有一些變化.分析圖1中的曲線及表2中的數據可以看出,化合物1-8的λ1、λ2和λ4三個吸收帶可歸于Salen部分;化合物1、2的λ3和λ5、λ6、λ7、λ8吸收帶可分別歸于卟啉部分的Soret帶和四個Q帶;化合物3-8的λ3和λ5、λ6吸收帶可分別歸于卟啉部分的Soret帶和兩個Q帶,化合物3、5、6、7、8的λ8吸收帶的弱電子吸收峰則可能是由于d-d躍遷所致及卟啉與Salen形成的共軛結構所產生的.
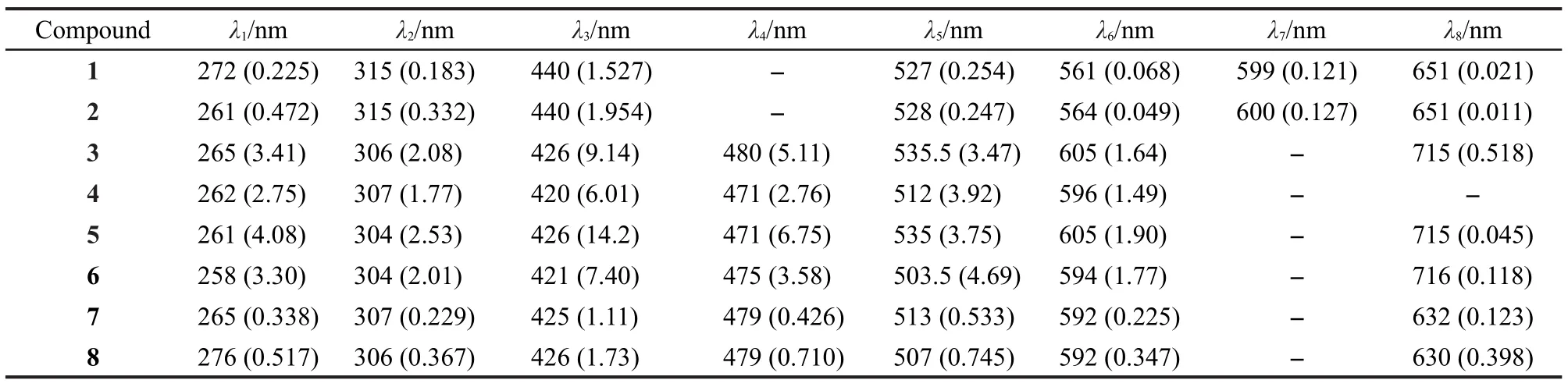
表2 化合物的紫外-可見光譜特征吸收峰數據aTable 2 UV-Vis absorbance data of compoundsa
3.4 熒光光譜
圖1為化合物A、2及其金屬鎳化合物3-6的熒光光譜圖.化合物A的激發波長為480 nm,化合物2的激發波長為440 nm,金屬鎳化合物3-6的激發波長為420 nm.激發狹縫為10 nm,發射狹縫為20 nm.
由熒光發射光譜圖可以看出,化合物A和2的氯仿溶液分別在716和724 nm處出現強發射峰,并且化合物2較化合物A的熒光光譜約有8 nm的紅移,這是由于3,5-二叔丁基水楊醛的醛基與化合物A中的胺基發生縮合反應后,取代基上的電子參與了整個體系的共軛大π鍵,增大了共軛體系,使整個卟啉共軛體系上的電子躍遷能級降低所致.43

圖1 化合物A、2及3-6在氯仿中的熒光發射光譜圖Fig.1 Fluorescence spectra of compoundsA, 2,and 3-6 in chloroform
卟啉的共軛體系中連有供電子取代基會使卟啉的熒光強度增加,反之,連有吸電子取代基則會使熒光強度明顯降低.供電子基中含有未成鍵電子,而未成鍵電子可以激發轉移到卟啉環上.由于未成鍵電子的電子云為不等性的sp3雜化,幾乎與卟啉環上的π軌道平行,因而未成鍵電子可以參與卟啉環π電子的共軛,即擴大其共軛體系.這樣就使得供電子基取代的卟啉化合物的吸收光譜和發射光譜波長比未取代的原卟啉化合物要長,熒光效率也有提高.當卟啉環外側連有吸電子基時,雖然吸電子基中也含有未成鍵電子,但是未成鍵電子并不參與卟啉環共軛π鍵.因為吸電子基中的未成鍵電子躍遷到π*鍵上時,其軌道與π*軌道的空間分布差異很大,幾乎不交疊,未成鍵電子要從空間的一個方向(即其本身的軌道)向另一方向(π*軌道)躍遷,這是一種“禁阻躍遷”(躍遷的幾率很小),致使其熒光減弱,或者說對熒光具有一定的抑制作用.44化合物A的結構中含有兩個具有供電子效應的―NH2,兩個―NH2上的未成鍵電子的電子云與卟啉環的π軌道形成共軛,使得化合物A比化合物2的熒光要強.
當金屬Ni2+離子與卟啉配體之間發生配位以后,測定其熒光發射光譜,均出現熒光淬滅現象.這是由于金屬鎳屬于過渡金屬,Ni3+、Ni+離子具有順磁性,順磁性金屬離子的第一激發單重態(S1)同第一激發三重態(T1)之間存在系間竄躍的能量衰減形式.由于順磁性粒子的存在,加強了體系中帶電分子、極性分子和可極化分子的不規則運動所產生的瞬間磁效應,這種磁效應的微擾可與電子自旋耦合,從而改變自旋方向,使系間竄躍顯著增大.同時這種微擾作用也會對分子結構造成影響,使得激發分子與溶劑分子相互作用增加,造成能量損失的增加.另外,由于鎳離子的激發態能級(m*)介于其S1和T1之間,發生了S1→m*→T1及S1→T1的非輻射躍遷,而此過程也造成了能量的損失.正是以上的多種原因造成了卟啉金屬鎳配合物熒光的淬滅.45
4 結論
采用1H NMR、紅外光譜、紫外-可見光譜、熒光及質譜等方法對Salen-卟啉型配體及單、雙核金屬配合物的研究表明,本文合成的目標化合物所包含的卟啉和Salen部分是共軛相連,整個分子具有π共軛骨架結構.金屬離子與分子中的兩個配位空腔的分別配位或雙配位,形成了單核鎳、雙核鎳和異雙核鎳、鋅金屬配合物.單核鎳及異雙核鎳、鋅配合物中,鎳離子落入Salen部分的配位空腔,而鋅離子則是與卟啉部分形成鋅卟啉大環結構.Salen-卟啉型配體的熒光由于金屬離子的配位而出現猝滅現象.
(1)Andruh,M.Chem.Commun.2007,No.25,2565.
(2) McInnes,E.J.L.;Piligkos,S.;Timco,G.A.;Winpenny,R.E.P. Coord.Chem.Rev.2005,249(23),2577.doi:10.1016/j.ccr. 2005.02.003
(3) Cooke,M.W.;Hanan,G.S.Chem.Soc.Rev.2007,36(9),1466. doi:10.1039/b609200b
(4) Beltran,L.M.C.;Long,J.R.Accounts Chem.Res.2005,38(4), 325.doi:10.1021/ar040158e
(5) Manoli,M.;Prescimone,A.;Bagai,R.;Mishra,A.;Murugesu, M.;Parsons,S.;Wernsdorfer,W.;Christou,G.;Brechin,E.K. Inorg.Chem.2007,46(17),6968.doi:10.1021/ic7007528
(6) Beletskaya,I.;Tyurin,V.S.;Tsivadze,A.Y.;Guilard,R.;Stern, C.Chem.Rev.2009,109(5),1659.doi:10.1021/cr800247a
(7) Jin,B.;Lee,H.M.;Lee,Y.A.;Ko,J.H.;Kim,C.;Kim,S.K. J.Am.Chem.Soc.2005,127(8),2417.doi:10.1021/ja044555w
(8) Balaban,T.S.Accounts Chem.Res.2005,38(4),612.
(9)Elemans,J.A.A.W.;Hameren,R.;Nolte,R.J.M.;Rowan,A. E.Adv.Mater.2006,18(10),1251.doi:10.1002/adma. 200502498
(10) Imahori,H.J.Phys.Chem.B 2004,108(20),6130.doi: 10.1021/jp038036b
(11)Zhang,X.H.;Jiao,Z.;Yan,W.W.;Ruan,W.J.;Zhu,Z.A.Acta Phys.-Chim.Sin.2010,26,701. [張曉紅,矯 志,閆偉偉,阮文娟,朱志昂.物理化學學報,2010,26,701.]doi:10.3866/ PKU.WHXB20100306
(12)Ren,X.F.;Ren,A.M.;Wang,Q.;Feng,J.K.Acta Phys.-Chim. Sin.2010,26,110.[任雪峰,任愛民,王 欽,封繼康.物理化學學報,2010,26,110.]doi:10.3866/PKU.WHXB20100103
(13) Song,C.E.;Lee,S.G.Chem.Rev.2002,102(10),3495.doi: 10.1021/cr0103625
(14) Katsuki,T.Synlett 2003,No.3,281.
(15)Venkataramanan,N.S.;Kuppuraj,G.;Rajagopal,S.Coord. Chem.Rev.2005,249(11-12),1249.doi:10.1016/j.ccr. 2005.01.023
(16) Jin,C.;Jia,Y.J.;Wang,B.J.;Fan,B.B.;Ma,J.H.;Li,R.F. Acta Phys.-Chim.Sin.2006,22,947.[晉 春,賈銀娟,王寶俊,范彬彬,馬靜紅,李瑞豐.物理化學學報,2006,22,947.] doi:10.3866/PKU.WHXB20060808
(17) Lu,Z.;Yuan,M.;Pan,F.;Gao,S.;Zhang,D.;Zhu,D.Inorg. Chem.2006,45(9),3538.doi:10.1021/ic051648l
(18) Lacroix,P.G.Eur.J.Inorg.Chem.2001,No.2,339.
(19) Binnemans,K.Chem.Rev.2005,105(11),4148.doi:10.1021/ cr0400919
(20) Liu,J.;Guo,L.Q.;Zhang,X.H.;Ruan,W.J.;Zhu,Z.A.Acta Phys.-Chim.Sin.2012,28,265. [劉 佳,郭莉芹,張曉紅,阮文娟,朱志昂.物理化學學報,2012,28,265.]doi:10.3866/ PKU.WHXB201111251
(21) Guo,L.Q.;Shi,X.L.;Ruan,W.J.;Zhang,X.H.;Zhu,Z.A. Acta Phys.-Chim.Sin.2010,26,1195.[郭莉芹,史秀麗,阮文娟,張曉紅,朱志昂.物理化學學報,2010,26,1195.]doi: 10.3866/PKU.WHXB20100336
(22) Leadbeater,N.E.;Marco,M.Chem.Rev.2002,102(10),3217. doi:10.1021/cr010361c
(23) Drain,C.M.;Varotto,A.;Radivojevic,I.Chem.Rev.2009,109 (5),1630.doi:10.1021/cr8002483
(24) Perrin,D.D.;Armarego,W.L.F.;Perrin,D.R.Purification of Laboratory Chemicals,2nd ed.;Chemical Laboratory Press: Beijing,1987;p 126;translated by Shi,Y. [Perrin,D.D.; Armarego,W.L.F.;Perrin,D.R.實驗室化學藥品的提純方法.第二版.時 雨,譯.北京:化學工業出版社,1987:126.]
(25) Kadish,K.M.;E,W.;Sintic,P.J.;Ou,Z.;Shao,J.;Ohkubo,K.; Fukuzumi,S.;Govenlock,L.J.;McDonald,J.A.;Try,A.C.; Cai,Z.L.;Reimers,J.R.;Crossley,M.J.J.Phys.Chem.B 2007,111(30),8762.doi:10.1021/jp0726743
(26)Thordarson,P.;Marquis,A.;Crossley,M.J.Org.Biomol.Chem. 2003,1(7),1216.doi:10.1039/b211015f
(27) Reek,J.N.;Rowan,A.E.;Crossley,M.J.;Nolte,R.J.J.Org. Chem.1999,64(18),6653.doi:10.1021/jo990348g
(28)Wang,C.Z.Study on Molecular Recognition,Asymmetric Catalysis and Theoretic Chemistry of Metalloporphyrins.Ph.D. Dissertation,Nankai University,Tianjin,2000.[王傳忠.金屬卟啉的分子識別、不對稱催化及理論研究[D].天津:南開大學,2000.]
(29)Wang,N.X.Nuclear Magnetic Resonance Spectroscopy; Chemical Industry Press:Beijing,2006;pp 15-25. [王乃興.核磁共振譜學.北京:化學化工出版社,2006:15-25.]
(30)Kruper,W.J.;Chamberlin,T.A.;Kochanny,M.J.Org.Chem. 1989,54(11),2753.doi:10.1021/jo00272a057
(31) Gouterman,M.J.Chem.Phys.1959,30(5),1139.doi:10.1063/ 1.1730148
(32)Adler,A.D.;Longo,F.R.;Finarelli,J.D.;Goldmacher,J.; Assour,J.;Korsakoff,L.J.Org.Chem.1967,32(2),476.
(33)Yeow,E.K.L.;Steer,R.P.Phys.Chem.Chem.Phys.2003,5 (1),97.
(34) Wang,D.;Zhang,J.;Shi,T.;Wang,B.;Cao,X.;Li,T. J.Photochem.Photobiol.A 1996,93(1),21.doi:10.1016/ 1010-6030(95)04142-7
(35) Gouterman,M.In The Porphyrins;Dolphin,D.Ed.;Academic Press:New York,1979;Vol.III,PartA,pp 1-156.
(36) Sacconi,L.;Ciampolini,M.;Maggio,F.;Cavasino,F.P.J.Am. Chem.Soc.1962,84(17),3246.doi:10.1021/ja00876a005
(37) Carlin,R.L.Transition Metal Chemistry;Marcel Dekker,New York,1965,p 239.
(38) Tas,E.;Aslanoglu,M.;Kilic,A.;Kara,Z.J.Coord.Chem. 2006,59(8),861.doi:10.1080/00958970500412206
(39) Odaba?o?lu,M.;Arslan,F.;?lmez,H.;Büyükgüng?r,O.Dyes Pigm.2007,75(3),507.doi:10.1016/j.dyepig.2006.06.033
(40) Chen,Z.;Wu,Y.;Gu,D.;Gan,F.Spectrochim.Acta Part A 2007,68(3),918.doi:10.1016/j.saa.2007.01.006
(41)Chen,Z.;Huang,F.;Wu,Y.;Gu,D.;Gan,F.Inorg.Chem. Commun.2006,9(1),21.doi:10.1016/j.inoche.2005.10.006
(42) Kasumov,V.T.Spectrochim.Acta Part A 2003,57(8),1649.
(43)Zhang,H.S.;Wang,H.;Zhao,Y.Y.Molecular Probes and Detection Reagents;Science Press:Beijing,2002;pp 25-98.[張華山,王 紅,趙媛媛.分子探針與檢測試劑.北京:科學出版社,2002:25-98.]
(44) Hariman,A.;Odaba?o?lu,M.;Arslan,F.;?lmez,H.; Büyükgüng?r,O.J.Chem.Soc.Faraday Trans.1 1981,77(2), 369.doi:10.1039/f19817700369
(45) Penzer,G.R.Molecular Luminescence Analytical Methods (Fluorescence and Phosphorescence Methods);Fudan University Press:Shanghai,1985;p 10;translated by Zhu,D. C.,Chen,J.H.,Zhu,S.S.[Penzer,G.R.分子發光分析法(熒光法和磷光法).祝大昌,陳劍宏,朱世盛,譯.上海:復旦大學出版社,1985:10.]
