大黃中4個(gè)番瀉苷類(lèi)成分受熱條件下轉(zhuǎn)化關(guān)系的研究
2012-07-26 11:35:14李瑞明張?zhí)m蘭閆希軍
中成藥 2012年11期
關(guān)鍵詞:實(shí)驗(yàn)
哈 飛, 李瑞明, 張?zhí)m蘭*, 閆希軍
(1.天津中醫(yī)藥大學(xué),天津300193;2.天津天士力集團(tuán)研究院現(xiàn)代中藥所,天津300410)
大黃為蓼科植物掌葉大黃Rheum palmatum L.、唐古特大黃Rheum tanguticum Maxim.ex Balf或藥用大黃Rheum officinale Baill.的干燥根及根莖[1],始載于《神農(nóng)本草經(jīng)》,列為中品,因其色黃而得名[2]。大黃是中醫(yī)中傳統(tǒng)的瀉劑之一,致瀉的主要成分為蒽醌類(lèi)化合物,其中以番瀉苷的作用最強(qiáng),游離型蒽醌瀉下作用較弱。番瀉苷A和B是大黃的活性成分,韓國(guó)及日本均將番瀉苷A作為檢測(cè)指標(biāo)[3]。自古研究大黃以來(lái),大黃有在處方中需要后下之說(shuō),藥典用法有不宜久煎,都是因?yàn)榧訜釙r(shí)間過(guò)長(zhǎng)瀉下成分會(huì)分解[4-5],但很少有文獻(xiàn)報(bào)道其具體成分轉(zhuǎn)化途徑,本實(shí)驗(yàn)對(duì)大黃中主要瀉下成分番瀉苷類(lèi)進(jìn)行了受熱條件下轉(zhuǎn)化關(guān)系的研究,闡述了其分解途徑,對(duì)大黃在中藥制劑中的應(yīng)用具有科學(xué)性指導(dǎo)。
1 儀器與試藥
1.1 儀器 Waters高效液相色譜儀 (Waters e2695 Separations Module,Waters 2998 PDA檢測(cè)器,Empower數(shù)據(jù)處理軟件),質(zhì)譜儀 (Agilent G6300 MS TRAP)調(diào)溫電熱套 (98-1-B型),電子分析天平 (梅特勒/XS205DU)。
1.2 試藥 大黃購(gòu)于安國(guó)藥材市場(chǎng),為四川大黃統(tǒng)貨,經(jīng)天津中醫(yī)藥大學(xué)李天祥教授鑒定為藥用大黃 Rheum officinale Baill.。
番瀉苷A、B、C、D對(duì)照品 (天津馬克生物技術(shù)有限公司,批號(hào):f-20091215),磷酸 (天津市光復(fù)精細(xì)化工研究所,色譜純),食用乙醇,二純水 (Milli-Q制備),色譜乙腈 (Merck公司)。
2 方法與結(jié)果
2.1 色譜條件 Agilent ZORBAX SB-C18色譜柱(4.6 mm×250 mm,5 μm);流動(dòng)相為乙腈 (A)-0.1%磷酸水溶液 (B)梯度洗脫,0~50 min(10% ~27%A),50~55 min(27% ~27%A),55~78 min(27% ~53%A),78~83 min(53%~80%A),83~93 min(80%A);柱溫35℃;檢測(cè)波長(zhǎng)280 nm;體積流量1.0 mL/min。
2.2 供試品溶液制備
2.2.1 大黃原液樣品制備 以最優(yōu)工藝[6]提取大黃生藥,稱(chēng)取大黃生藥20.0 g,加入10倍生藥量的80%乙醇,回流提取3次,每次提取時(shí)間為0.5 h,得大黃提取液。取樣,為0時(shí)刻待測(cè)液。分別平行取6份大黃提取液,每份200 mL置于圓底燒瓶中,回流加熱,回流時(shí)間依次為10、20、30、40、50、60 min。分別取樣平行3份,以2.1項(xiàng)色譜條件方法檢測(cè),比較大黃中4個(gè)番瀉苷類(lèi)成分變化。
2.2.2 番瀉苷樣品制備 分別精密稱(chēng)取番瀉苷A、B、C、D各2.5 mg,用80%乙醇定容于25 mL量瓶中,得0.1 mg/mL供試品溶液。取樣,為0時(shí)刻待測(cè)液。分別將供試品溶液移置50 mL燒瓶中,電熱套加熱回流,分別在10、20、30、40、50、60 min時(shí)刻取樣,得待測(cè)液,以2.1項(xiàng)色譜條件方法檢測(cè)。
2.3 方法學(xué)考察
2.3.1 線性 分別精密吸取番瀉苷A、B、C、D對(duì)照品原溶液3、5、7、9、12、15 μL,按2.1項(xiàng)色譜條件,高效液相檢測(cè),測(cè)得峰面積為縱坐標(biāo),進(jìn)樣量為橫坐標(biāo),回歸方程分別為:YA=862384 X+421483,r=0.9995;YB=919823X -28414,r=0.9990;YC=541435X -12076,r=0.9991;YD=929651X -40147,r=0.9998。番瀉苷A、B、C、D 在0.3 μg~1.5 μg范圍內(nèi)呈良好線性關(guān)系。
2.3.2 精密度實(shí)驗(yàn) 分別精密吸取番瀉苷A、B、C、D對(duì)照品原溶液10 μL,按2.1項(xiàng)色譜條件下重復(fù)進(jìn)樣6次,測(cè)得各峰面積,得其RSD值分別為0.26%、0.37%、0.45%和0.43%,表明精密度良好。
2.3.3 重復(fù)性實(shí)驗(yàn) 供試品溶液每時(shí)刻分別平行取樣3份,進(jìn)行測(cè)定,得番瀉苷A、B、C、D峰面積值的RSD均小于3%,表明重復(fù)性良好。
2.3.4 穩(wěn)定性實(shí)驗(yàn) 取番瀉苷A、B、C、D對(duì)照品原溶液,分別于0、2、4、6、8、10、12、24 h在2.1項(xiàng)色譜條件下進(jìn)行測(cè)定,峰面積的RSD值分別為1.06%、1.27%、0.95%和1.31%,表明在常溫下24 h內(nèi)穩(wěn)定。
2.4 結(jié)果
2.4.1 大黃提取液未加熱前HPLC色譜圖見(jiàn)圖1。大黃中番瀉苷A、B、C、D成分在受熱條件下均存在降解,通過(guò)對(duì)大黃中番瀉苷A、B、C、D在受熱條件下各時(shí)間點(diǎn)的相對(duì)峰面積進(jìn)行比對(duì),分別得到1 h內(nèi)的變化趨勢(shì),見(jiàn)圖2~5。
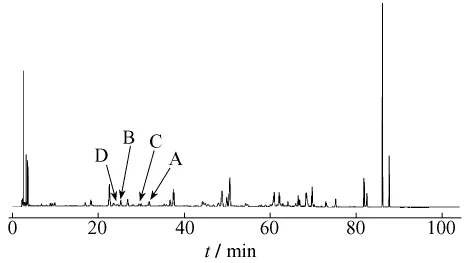
圖1 大黃提取液HPLC圖譜Fig.1 Chromatogram of rhubarb extract
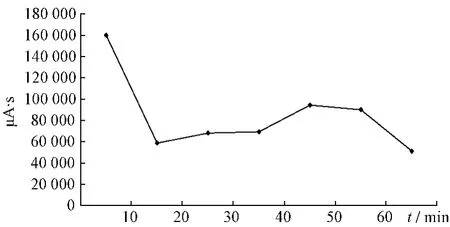
圖2 番瀉苷A變化趨勢(shì)圖Fig.2 Changed trend chart of sennoside A
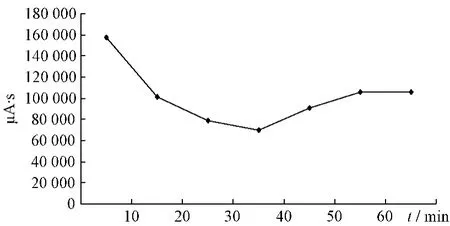
圖3 番瀉苷B變化趨勢(shì)圖Fig.3 Changed trend chart of sennoside B
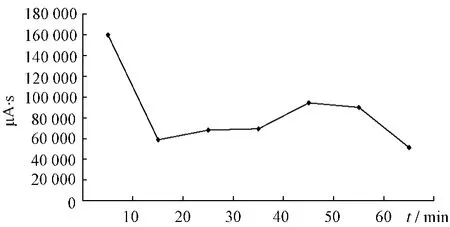
圖4 番瀉苷C變化趨勢(shì)圖Fig.4 Changed trend chart of sennoside C
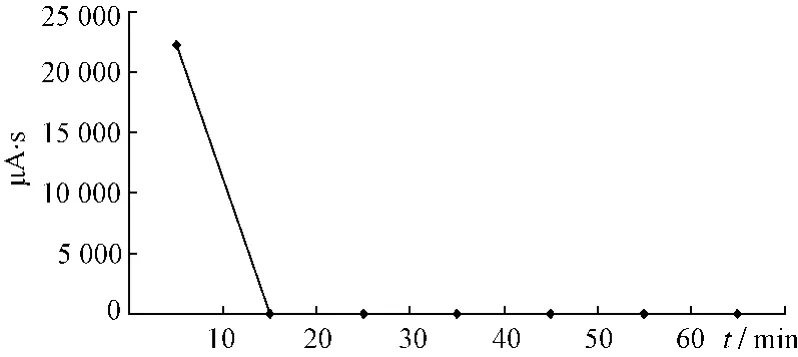
圖5 番瀉苷D變化趨勢(shì)圖Fig.5 Changed trend chart of sennoside D
2.4.2 番瀉苷A、B、C、D在受熱條件下存在相互轉(zhuǎn)化關(guān)系,在1 h內(nèi)呈規(guī)律性趨勢(shì)變化,結(jié)果見(jiàn)圖6~9。
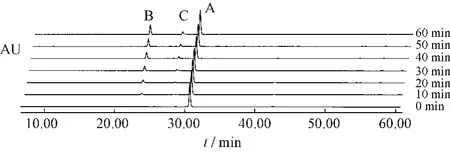
圖6 番瀉苷A受熱條件下1h內(nèi)變化情況Fig.6 Sennoside A changes under heating condition in 1 h
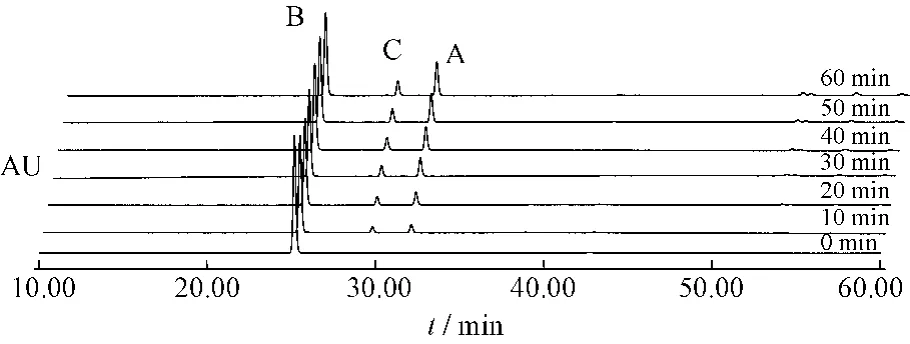
圖7 番瀉苷B受熱條件下1 h內(nèi)變化情況Fig.7 Sennoside B changes under heating condition in 1 h
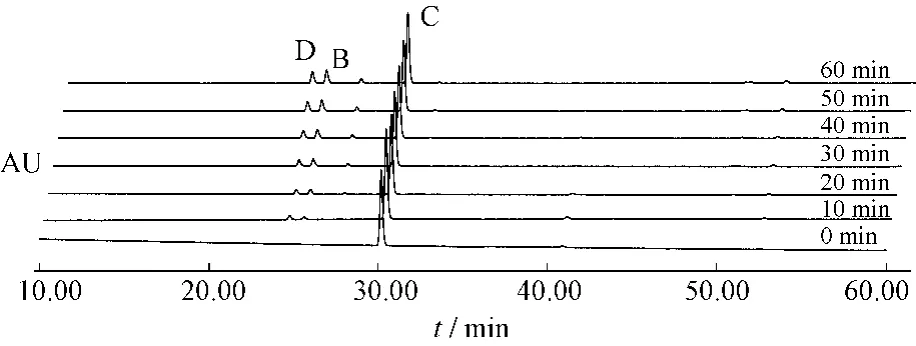
圖8 番瀉苷C受熱條件下1 h內(nèi)變化情況Fig.8 Sennoside C changes under heating condition in 1 h
番瀉苷A在1 h內(nèi)80%乙醇中加熱回流條件下,可以轉(zhuǎn)化為番瀉苷B和番瀉苷C。番瀉苷B在1 h內(nèi)80%乙醇中加熱回流條件下,可以轉(zhuǎn)化為番瀉苷A和番瀉苷C。番瀉苷C在1 h內(nèi)80%乙醇中加熱回流條件下,可以轉(zhuǎn)化為番瀉苷B和番瀉苷D和新的中間產(chǎn)物。番瀉苷D在1 h內(nèi)80%乙醇中加熱回流條件下,可以轉(zhuǎn)化為番瀉苷B和番瀉苷C和新的中間產(chǎn)物。見(jiàn)表1~4。
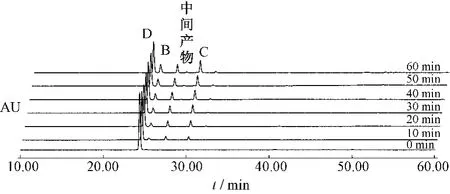
圖9 番瀉苷D受熱條件下1 h內(nèi)變化情況Fig.9 Sennoside D changes under heating condition in 1 h
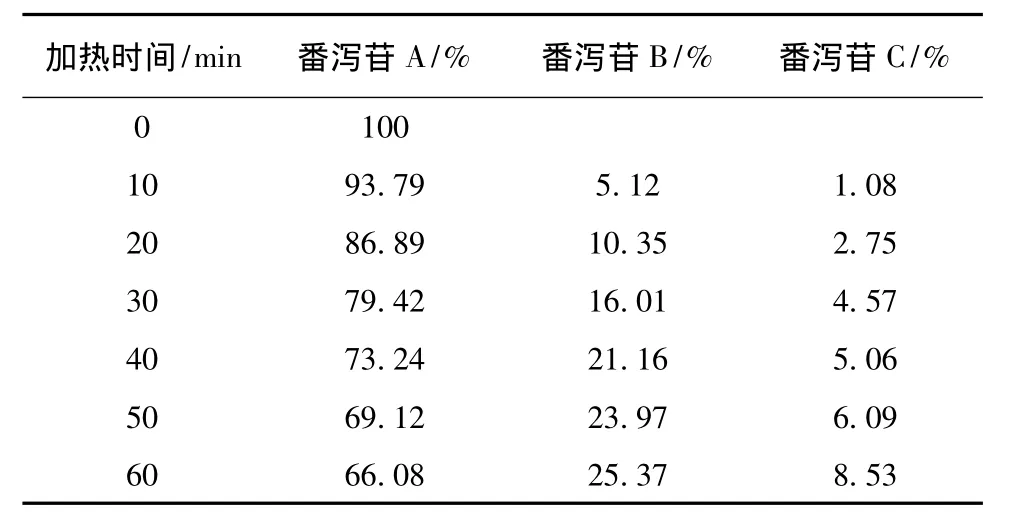
表1 番瀉苷A在加熱回流各時(shí)間點(diǎn)的峰面積百分比Tab.1 Peak area percentage of sennoside A under heating reflux condition each time
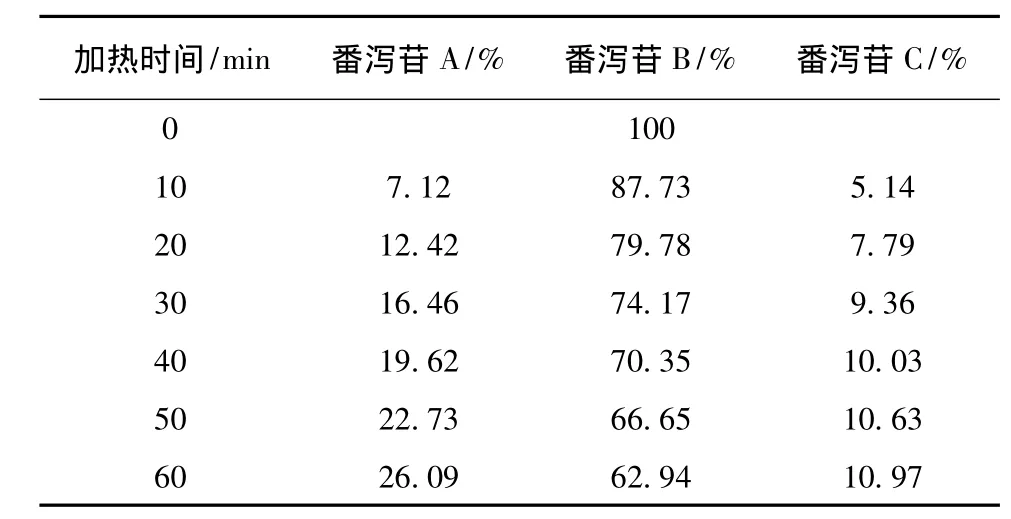
表2 番瀉苷B在加熱回流各時(shí)間點(diǎn)的峰面積百分比Tab.2 Peak area percentage of sennoside A under heating reflux condition each time
番瀉苷A、B、C、D化學(xué)結(jié)構(gòu)式[7]見(jiàn)圖10。
番瀉苷A和B互為異構(gòu)體,相對(duì)分子質(zhì)量為862.74;番瀉苷C和D互為異構(gòu)體,相對(duì)分子質(zhì)量為848.74[8]。取番瀉苷D加熱60 min時(shí)刻的樣品,進(jìn)行質(zhì)譜檢測(cè)。番瀉苷C和D加熱下有新中間產(chǎn)物生成,對(duì)其進(jìn)行質(zhì)譜分析,對(duì)正離子碎片離子峰采集,結(jié)果如圖11,得知推測(cè)生成的中間產(chǎn)物的相對(duì)分子質(zhì)量為834.7。
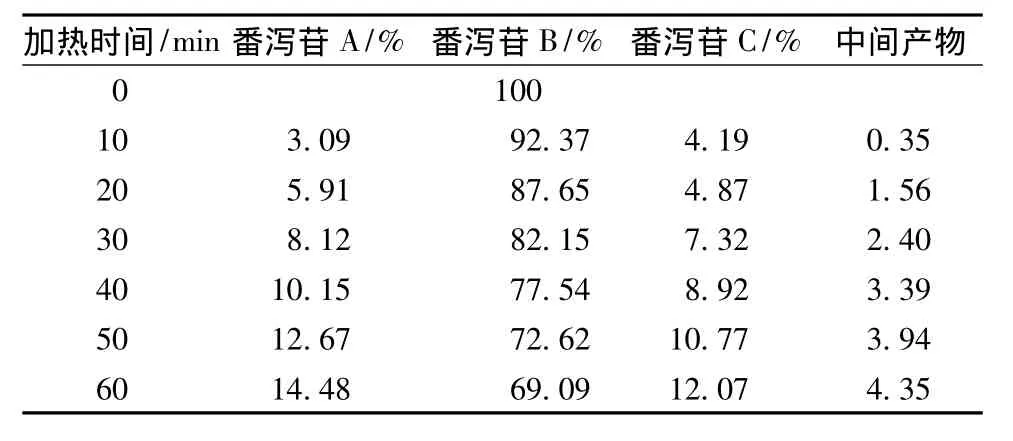
表3 番瀉苷C在加熱回流各時(shí)間點(diǎn)的峰面積百分比Tab.3 Peak area percentage of sennoside A under heating reflux condition each time
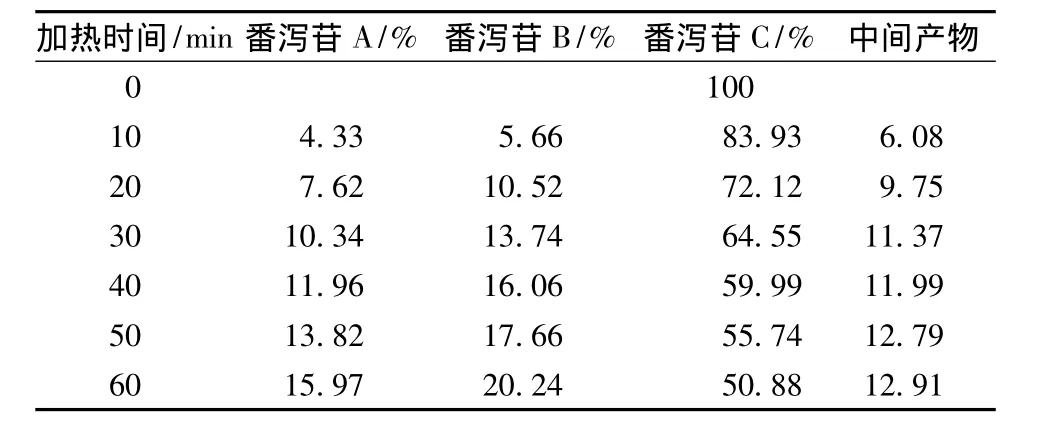
表4 番瀉苷D在加熱回流各時(shí)間點(diǎn)的峰面積百分比Tab.4 Peak area percentage of sennoside A under heating reflux condition each time
3 討論
大黃瀉下的有效成分為蒽醌類(lèi)衍生物,以蒽酮苷類(lèi)瀉下效力較強(qiáng),游離蒽酮較弱,游離蒽醌更次。雙蒽酮比單蒽酮強(qiáng),其中番瀉苷A(sennoside A)為大黃瀉下最強(qiáng)的有效成分[9]。因番瀉苷A加熱分解,大黃煎煮時(shí)間過(guò)長(zhǎng),會(huì)使瀉下作用減弱[10]。大黃中番瀉苷類(lèi)成分為大黃瀉下作用的主要成分[11],藥效作用大,但含有量較少,因此有必要對(duì)番瀉苷類(lèi)單體成分進(jìn)行研究,使大黃更好的在現(xiàn)代中藥中應(yīng)用。
本實(shí)驗(yàn)中,對(duì)大黃中番瀉苷類(lèi)成分受熱條件下考察,番瀉苷A、B、C、D成分均有降解,為明確其降解原因,故首次將番瀉苷A、B、C、D對(duì)照品作為考察對(duì)象,更直觀的對(duì)大黃中番瀉苷類(lèi)成分穩(wěn)定性進(jìn)行研究,揭示了番瀉苷類(lèi)成分之間的轉(zhuǎn)化關(guān)系,避免了大黃中其他成分對(duì)番瀉苷類(lèi)成分研究時(shí)的影響。
大黃藥材中番瀉苷總量在0.304%~1.450%之間,均值在0.892%[12],其中含番瀉苷D較低,同時(shí),從單體番瀉苷D受熱下轉(zhuǎn)化情況得知其降解率大,且轉(zhuǎn)化中難形成番瀉苷D,推測(cè)為大黃中番瀉苷D受熱全部降解的原因,HPLC未能檢測(cè)出。
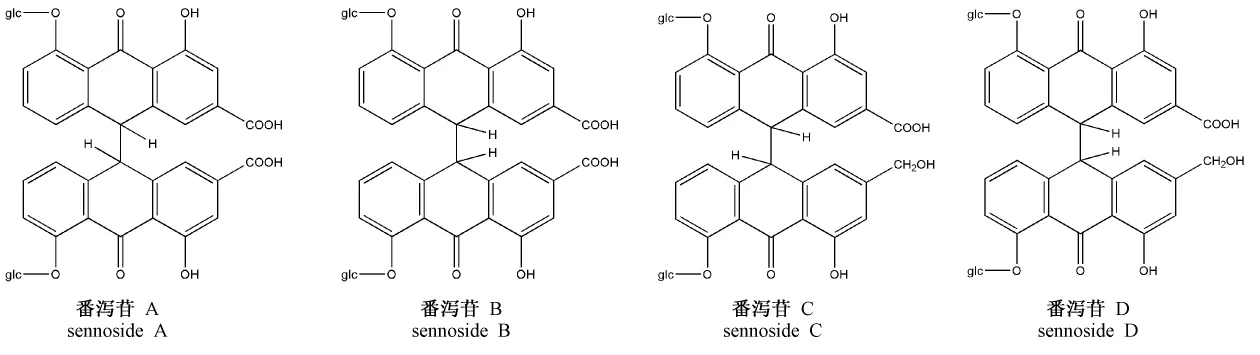
圖10 番瀉苷A、B、C、D化學(xué)結(jié)構(gòu)式Fig.10 Chemical structures of sennoside A,B,C,D

圖11 中間產(chǎn)物質(zhì)譜圖譜Fig.11 Mass spectrum of intermediate product
結(jié)合型蒽醌以番瀉苷為主,是大黃蒽醌類(lèi)衍生物以及蒽酮與葡萄糖結(jié)合形成的苷類(lèi)。蒽酮糖苷進(jìn)行治療時(shí)發(fā)揮作用的成分為水解后苷元的部分[13]。大黃中番瀉苷類(lèi)成分的致瀉作用是因其在腸內(nèi)變?yōu)榇簏S酸蒽酮所致,并進(jìn)一步被氧化成番瀉苷元[7,14]。結(jié)合實(shí)驗(yàn)結(jié)果推測(cè),二蒽酮番瀉苷類(lèi)成分在受熱條件下可斷鏈形成單蒽酮,又可再結(jié)合,形成互相轉(zhuǎn)化過(guò)程。
本實(shí)驗(yàn)考察番瀉苷類(lèi)成分是為大黃更好的應(yīng)用為前提,大黃提取醇提法也為經(jīng)典法[15],故只考慮了80%醇中和受熱條件下其轉(zhuǎn)化關(guān)系。對(duì)于番瀉苷類(lèi)成分在不同溶媒及不同pH和不同溫度條件下的變化關(guān)系,有待進(jìn)一步研究。
實(shí)驗(yàn)中,在質(zhì)譜分析中,番瀉苷D在加熱條件下生成的新中間產(chǎn)物量較大,所以選擇其加熱60 min時(shí)刻進(jìn)行分析。番瀉苷A和B的相對(duì)分子質(zhì)量已知為862.74,番瀉苷C和D的相對(duì)分子質(zhì)量為848.74,故通過(guò)質(zhì)譜正離子譜圖得其中間產(chǎn)物分子量。
該實(shí)驗(yàn)對(duì)大黃中4個(gè)番瀉苷類(lèi)成分的穩(wěn)定性進(jìn)行研究,數(shù)據(jù)真實(shí)可靠,為大黃在中藥現(xiàn)代化中的應(yīng)用提供可靠性指導(dǎo)。
[1]國(guó)家藥典委員會(huì).中華人民共和國(guó)藥典:2010年版一部[S].北京:中國(guó)醫(yī)藥科技出版社,2010:22.
[2]張向紅,程黎暉.大黃的藥理作用及臨床應(yīng)用研究進(jìn)展[J].中國(guó)藥業(yè),2009,18(21):76-78.
[3]馬 蓉,張雪菊.液相色譜法對(duì)不同產(chǎn)地大黃中番瀉苷A含量比較分析[J].中成藥,2008,30(10):1489-1490.
[4]馮 萍,趙 萍.劑量、炮制和煎服方法對(duì)大黃藥效的影響[J].實(shí)用中醫(yī)內(nèi)科雜志,2004,18(3):256-258.
[5]朱詩(shī)塔,雷 鵬,李新中,等.掌葉大黃不同炮制品瀉下、止血作用的比較研究[J].中藥材,2008,31(2):199-201.
[6]哈 飛,李瑞明,張?zhí)m蘭.大黃濃縮干燥過(guò)程中各活性成分的穩(wěn)定性[J]中國(guó)實(shí)驗(yàn)方劑學(xué)雜志,2012,18(3):11-13.
[7]吳立軍.天然藥物化學(xué)[M].5版.北京:人民衛(wèi)生出版社:148-149.
[8]邱頌平.大黃的藥學(xué)與臨床研究[M].北京:中國(guó)中醫(yī)藥出版社,2007.12:143-145.
[9]李 娟,李 堅(jiān).大黃藥理作用研究及臨床應(yīng)用概況[J].實(shí)用醫(yī)藥雜志,2006,23(9):1132-1133.
[10]岡村信幸.制備大黃煎劑時(shí)番瀉苷A及瀉下活性的變化[J].國(guó)外醫(yī)學(xué)中醫(yī)中藥分冊(cè),2003,25(2):98-99.
[11]鄭志華,祝 晨.HPLC測(cè)定大黃提取工藝產(chǎn)物番瀉苷A的含量[J].中藥材,2004,27(12):950-951.
[12]孫 佩,李 敏.HPLC法測(cè)定大黃藥材和飲片中番瀉苷A和番瀉苷B的含量[J].成都中醫(yī)藥大學(xué)學(xué)報(bào),2008,31(3):51-23.
[13]王 磊,高文遠(yuǎn).大黃活性成分藥代動(dòng)力學(xué)研究進(jìn)展[J].中成藥,2011,33(9):1571-1573.
[14]武玉清,王靜霞,周成華,等.番瀉苷對(duì)小鼠腸道運(yùn)動(dòng)功能的影響及相關(guān)機(jī)制研究[J].中國(guó)臨床藥理學(xué)與治療學(xué),2004,9(2):162-164.
[15]馬 靜,張 建,劉春華,等.大黃浸膏制備工藝研究[J].中成藥,2011,33(3):518-521.
猜你喜歡
作文·小學(xué)低年級(jí)(2025年2期)2025-02-13 00:00:00
小雪花·小學(xué)生快樂(lè)作文(2024年11期)2024-12-31 00:00:00
作文·小學(xué)低年級(jí)(2024年2期)2024-04-29 00:00:00
作文·小學(xué)低年級(jí)(2023年3期)2023-04-29 00:00:00
小獼猴智力畫(huà)刊(2022年9期)2022-11-04 02:31:42
小主人報(bào)(2022年4期)2022-08-09 08:52:06
中學(xué)生數(shù)理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學(xué))(2019年6期)2019-10-10 01:01:50
發(fā)明與創(chuàng)新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
