銀苦黃白口服液的不同配伍對苦參堿和氧化苦參堿影響
2012-07-26 11:35:18李立順錢晶晶時維靜
中成藥 2012年11期
關鍵詞:中藥
李立順, 錢晶晶, 時維靜
(安徽科技學院,安徽鳳陽233100)
銀苦黃白口服液為臨床經驗方研制,由金銀花、苦參、黃芪、白茅根按6∶7∶7∶10組成。具有清熱祛濕,利水的功效,臨床用于腎炎、腎盂腎炎、尿毒癥等療效顯著。方中苦參系豆科植物苦參Sophora flavescens Air.的干燥根[1],具有清熱燥濕,利水的功效,其主要活性成分是苦參堿、氧化苦參堿[2]。銀苦黃白口服液采用的是水提醇沉制備工藝,為考察共煎對方中化學成分的影響,本試驗采用L8(27)正交設計,考察不同配伍對苦參堿、氧化苦參堿含有量的影響,以探討該方的配伍化學變化規律,為制備工藝的改進提供研究數據。
1 儀器與試藥
1.1 儀器 CAMAG LINOMAT5半自動點樣儀;CAMAG TLC Scanner 3薄層色譜掃描儀;winCATS軟件,瑞士卡瑪;CP225D準微量天平,德國賽多利斯;RE52-98旋轉蒸發儀,上海亞榮生化儀器廠;LG10-24A離心機,北京醫用離心機廠。
1.2 試藥 黃芪 Astragalus membranaceua(Fisch.)Bge.var.mongholicus(Bge.)Hsiao.產地內蒙古;白茅根 Imperata cylindrical Beauv.Var.major C.E.Hubb.產地安徽;金銀花Lonicera japonica Thunb.產地河南;苦參Sophora flavescens Ait.產地安徽。均購自安徽京皖中藥飲片廠,藥品生產許可證號:皖Y20020003批號20100319。經時維靜教授鑒定為正品。
苦參堿 (110805-200306)、氧化苦參堿 (0780-200004),購自中國藥品生物制品檢定所;硅膠薄層板G,20 cm×10 cm,購自青島海洋化工廠,批號20070724;試劑均為分析純。
2 方法與結果
2.1 L8(27)配伍的正交設計 選黃芪、金銀花、白茅根作為3個因素,選用藥和不用藥為2個水平,同時考慮兩兩交互作用,因素水平見表1。

表1 因素水平
2.2 供試溶液制備 按正交試驗設計配比稱取各味藥材,加8倍量水浸30 min,煎提2次,每次1 h,合并濾液,靜置12 h,取上清液。濃縮至每1 mL相當于1.5 g藥材,加乙醇使含醇量達75%,靜置,上清液回收乙醇,精濾,調整溶液量,使苦參含量均達35%,如法制備缺苦參樣品 (見表2)。精密吸取各樣品1 mL,過短中性氧化鋁柱(100~200目2 g,內徑1 cm),以50%甲醇洗脫約10 mL,洗脫液水浴蒸干后用甲醇溶解并定容至2 mL,冷藏備用。
2.3 對照品溶液的制備 精密稱取苦參堿、氧化苦參堿適量,分別置10 mL量瓶中,甲醇溶解至刻度,制成質量濃度為0.2 mg/mL的溶液。

表2 苦參不同配伍的用藥比例
2.4 薄層色譜條件 硅膠G板,板厚約0.3 mm;反復試驗后,選定三氯甲烷-甲醇-濃氨試液 (10∶1.2∶0.2)為展開劑,置雙槽展開缸,飽和20 min。取缺苦參、苦參藥材、全方及苦參堿、氧化苦參堿溶液,半自動點樣儀噴霧條帶點樣各2 μL,帶寬6 mm,各2重復。置雙槽展開缸的另側預平衡15 min,上行展開8 cm,取出吹干,噴改良碘化鉍鉀試液 (碘化鉍鉀試液1 mL,加0.6 moL/L鹽酸溶液2 mL,加水10 mL)顯色劑,至顯色清晰。室溫15℃,相對濕度40%。此條件下,苦參堿、氧化苦參堿顯橘黃色斑點,分離較好,可同板比較,缺苦參樣品無干擾。見圖1。
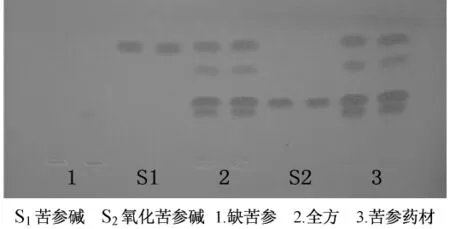
圖1 苦參及全方薄層圖譜
2.5 薄層掃描條件 使用薄層掃描儀,全光譜掃描,苦參堿、氧化苦參堿顯色后,最佳檢測波長λ=530 nm;采用吸收檢測 (光源為鎢燈),單波長反射直線掃描法,狹縫3.0 mm×0.45 mm,掃描速度20 mm/s。
2.6 線性關系考察 精密吸取苦參堿、氧化苦參堿對照品各1、2、3、4、5 μL,各重復2次,點于同一硅膠G薄層板上,按2.4項展開、顯色,并按2.5項測定峰面積積分值,以對照品量為橫坐標,相應的峰面積積分值為縱坐標,進行線性回歸。二者的回歸方程、相關系數及線性范圍分別為:苦參堿的回歸方程為 Y=56.801+1.409X,r=0.99867,苦參堿點樣量在0.20~1.00 μg范圍內呈良好的線性關系;氧化苦參堿的回歸方程為Y=2374.410+13.580X,r=0.99996,氧化苦參堿點樣量在0.20~1.00 μg范圍內呈良好的線性關系。
2.7 穩定性考查 對一薄層板的同一斑點每間隔2h掃描測定1次,結果顯示斑點在10 h內穩定,RSD為1.7%(n=5)。
2.8 精密度考查
2.8.1 同板精密度 精密吸取苦參堿、氧化苦參堿對照品溶液2 μL,于同一硅膠G薄層板上連續點5次,按2.4項展開、顯色,并按2.5項測定峰面積積分值。結果苦參堿RSD為1.27%(n=5);氧化苦參堿RSD為0.51%(n=5)。
2.8.2 異板精密度 精密吸取苦參堿、氧化苦參堿對照品溶液2 μL,分別點于5塊硅膠G薄層板上,按2.4項展開、顯色,并按2.5項測定峰面積積分值。結果苦參堿RSD為2.19%(n=5);氧化苦參堿RSD為1.88%(n=5)。
結果表明該方法的同板精密度、異板精密度均較好。
2.9 加樣回收率試驗 精密稱取已知含有量樣品9份,分別精密加入適量苦參堿、氧化苦參堿對照品,按供試品溶液制備法制備,按上述薄層色譜條件及薄層掃描條件測定供試品溶液中苦參堿、氧化苦參堿的含有量。結果苦參堿平均加樣回收率為98.67%,RSD為3.67%(n=9);氧化苦參堿的平均加樣回收率為99.55%,RSD為1.94%(n=9)。2.10 配伍對苦參堿和氧化苦參堿含有量的影響 精密吸取表2中1-8號供試品溶液各2 μL,苦參堿1 μL、5 μL,氧化苦參堿1 μL、6 μL進樣,各重復2次。按2.4項同板點樣、展開、顯色,并按2.5項測定峰面積積分值。采用線性回歸二點法計算含有量。測定不同配伍樣品苦參堿和氧化苦參堿的含有量見表3,方差分析見表4、表5。

表3 L8(27)正交試驗設計及結果

表4 苦參堿方差分析

表5 氧化苦參堿方差分析
從表3、4、5可以看出,苦參單味藥中苦參堿含有量最低,氧化苦參堿含有量最高。5號組 (金銀花、白茅根、苦參)苦參堿含有量最高。方差分析表明,黃芪與金銀花的交互作用對苦參堿含有量的影響具有顯著性差異 (P<0.05),即黃芪與金銀花共用或均不用苦參堿含有量較低;黃芪對氧化苦參堿含有量的影響具有顯著性差異 (P<0.05),白茅根對氧化苦參堿含有量的影響具有極顯著性差異 (P<0.01);但當白茅根與黃芪共同配伍苦參使用,明顯降低了氧化苦參堿的含有量。
3 討論
銀苦黃白湯是臨床治療腎炎、腎盂腎炎、尿毒癥的有效方。中藥復方用藥是中醫臨床治病的特色與靈魂[3]。方劑的藥效并不是單味藥效的簡單加和,而是遵循“君臣佐使”原則。強調方證關聯、病癥結合,使各味藥形成“有制之師”,針對相應的證或病,達到“整體綜合調節”的效果[4]。中藥復方治療疾病的物質基礎是其中的化學物質。在改劑型的過程中,只有明確其化學成分及其理化性質,明確中藥有效成分的化學結構,才有可能明確其制備過程的動態變化,才有可能實現中藥質量的穩定性和臨床療效的確切性,才能保證中藥復方制劑等生產過程的質量控制和檢測[5]。
本項試驗研究了銀苦黃白口服液不同配伍對苦參堿和氧化苦參堿含有量的影響。與配伍共煎相比苦參單味藥中苦參堿含有量最低,氧化苦參堿含有量最高,表明共煎不僅是增加了苦參堿的提取率,也有氧化苦參堿轉化為苦參堿可能[6]。3、4、5、6號共煎液中苦參堿和氧化苦參堿的總量高于單味苦參提取液的總量,其中5號 (苦參+銀花+白茅根) >4號 (苦參+黃芪) >6號 (苦參+銀花)>3號 (苦參+黃芪+白茅根),提示苦參與銀花、白茅根共煎能顯著增加苦參堿和氧化苦參堿的提取率;黃芪與金銀花共用或均不用組苦參堿含有量較低,提示黃芪與金銀花增加苦參堿提取率的途徑不同,共用時反而互相抑制,影響了苦參堿的提取;白茅根與黃芪共同配伍苦參使用,明顯降低了氧化苦參堿的含有量。從對苦參堿和氧化苦參堿含有量的影響角度,可以考慮黃芪不宜共煎。但對其他有效成分的影響及臨床療效的影響,還將進一步研究,另文報導。
試驗對比了多種展開劑,如苯-丙酮-醋酸乙酯-濃氨(2∶3 ∶4 ∶0.2)[7],甲苯-丙酮-甲醇 (8 ∶3 ∶0.5),甲苯-乙酸乙酯-甲醇-水 (2 ∶4 ∶2 ∶1)[1,8],環己烷-乙酸乙酯-丙酮-濃試液 (4 ∶6 ∶8 ∶0.5)[9],三氯甲烷-甲醇-水 (7 ∶2 ∶1)和三氯甲烷-甲醇 (5 ∶0.2)[10],三氯甲烷-甲醇-濃氨試液 (5∶0.6∶0.3)等。因苦參堿和氧化苦參堿Rf值相差較大,有的對苦參堿展開效果較好,有的對氧化苦參堿展開效果較好,經調整選定三氯甲烷-甲醇-濃氨試液 (10∶1.2∶0.2)為展開劑,可同板檢測兩項指標。噴改良碘化鉍鉀試液為顯色劑,顯色穩定。因CAMAG薄層色譜掃描儀具有自動扣除背景的作用,故選用單波長反射直線掃描,簡單、快速、準確。
[1]國家藥典委員會.中華人民共和國藥典:2010年版一部[S].北京:中國醫藥科技出版社,2010:188.
[2]李 毅,王 飛,吳 民.苦參藥材質量標準研究[J].中成藥,2011,33(2):363-364.
[3]賀福元,鄧凱文,石繼連,等.中藥有效成分群組方技術研究若干基礎瓶頸問題及數理特征化思路的提出[J].中藥材,2009,32(1):1-7.
[4]賀福元,鄧凱文,鄒 歡,等.中藥復方譜動學與譜效動力學差異性的研究[J].中國中藥雜志,2011,36(2):136-141.
[5]王智民.中藥藥效物質基礎的系統研究是中藥現代化的關鍵[J].中國中藥雜志,2003,28(12):1111-1113.
[6]萬旭英,羅 明,賀 平,等.苦參堿和氧化苦參堿的提取鑒定和含量測定[J].中華中醫藥學刊,2009,27(10):2066-2068.
[7]姜志戎,李國柱,陳 明.婦潔洗劑的質量標準研究[J].中成藥,2009,31(8):14-15.
[8]王惠娟,楊國亮,麻風華,等.苦參飲片質量標準研究[J].中國藥房,2010,21(23):2151-2153.
[9]祁海宏,鄔瑾麗,屈鵬舒,等.薄層色譜掃描法測定治帶片中苦參堿含量[J].中國藥業,2008,17(3):15-16.
[10]丁郁文.苦參薄層色譜法的改進[J].陜西中醫,2008,29(8):1071-1072.
猜你喜歡
中老年保健(2021年5期)2021-12-02 15:48:21
中老年保健(2021年4期)2021-12-01 11:19:40
中老年保健(2021年4期)2021-08-22 07:08:32
中國現代中藥(2020年10期)2020-12-16 08:53:18
金橋(2020年7期)2020-08-13 03:07:00
基層中醫藥(2020年12期)2020-07-22 06:34:38
中國現代中藥(2020年4期)2020-06-10 09:56:34
基層中醫藥(2018年6期)2018-08-29 01:20:20
長春中醫藥大學學報(2017年1期)2017-04-16 05:56:49
肝博士(2015年2期)2015-02-27 10:49:49
