高效陰離子交換色譜-脈沖安培法檢測低聚異麥芽糖
2012-09-12 13:22:20張曉萍段鋼
食品與發(fā)酵工業(yè) 2012年7期
關鍵詞:分析
張曉萍,段鋼
(杰能科(中國)生物工程有限公司,江蘇無錫,214028)
高效陰離子交換色譜-脈沖安培法檢測低聚異麥芽糖
張曉萍,段鋼
(杰能科(中國)生物工程有限公司,江蘇無錫,214028)
建立了高效陰離子交換色譜-脈沖安培檢測法定量分析低聚異麥芽糖的方法。采用CarboPakTMPA10色譜柱,配合安培檢測器,以NaOH及醋酸鈉為洗脫劑。采用此方法不僅一次性實現(xiàn)了低聚異麥芽糖常規(guī)組分中葡萄糖、麥芽糖、麥芽三糖、異麥芽糖、異麥芽三糖,潘糖的有效分離,也實現(xiàn)了異麥芽四糖、異麥芽五糖、異麥芽六糖、異麥芽七糖、海藻糖、麥芽酮糖、曲二糖、黑曲霉糖高聚合度糖及二糖同分異構體間的分離及檢測。以不同濃度的標準糖混合溶液建立了校正曲線,此方法中,各組分在0.032~25.975 mg/L間具有良好的線性關系,各物質的檢測限和定量限分別在0.008~0.022 mg/L和0.027~0.073 mg/L,樣品加標回收率為82.02%~116.37%。
低聚異麥芽糖,同分異構體,分離,高效陰離子交換色譜-脈沖安培法
低聚異麥芽糖是目前市場上最主要的一種功能性低聚糖,具有難消化性及選擇性增殖雙歧桿菌及乳酸桿菌從而維持腸道微生態(tài)平衡,促進人體健康的功能[1]。低聚異麥芽糖是淀粉經(jīng)過液化、糖化、轉苷等工序制備得到,產(chǎn)品中的有效成分主要指異麥芽糖、潘糖、異麥芽三糖及聚合度大于等于4的糖[2]。在低聚異麥芽糖的酶法制備過程中,由于所使用的轉苷酶的作用特性,使得其產(chǎn)物不僅有異麥芽糖、潘糖及異麥芽三糖這些主要的有效成分,還會得到其它的轉苷產(chǎn)物,如曲二糖、黑曲霉糖等異構體。由于異麥芽糖、麥芽糖、曲二糖及黑曲霉糖屬于2個葡萄糖分子通過不同糖苷鍵連接所形成的位置異構體,因此它們的結構極其相似,目前常用的分析方法并不能實現(xiàn)這些同分異構體的有效分離。
目前分析低聚異麥芽糖的方法主要是高效液相色譜法,分為只使用氨基鍵合柱的單柱法和使用離子交換柱和氨基鍵合柱的雙柱法。單柱法中所使用的氨基鍵合柱能夠實現(xiàn)異麥芽糖、麥芽糖、潘糖、麥芽三糖及異麥芽三糖間的分離,但此種類型的色譜柱對聚合度較高的糖具有吸附作用[3],從而使分析結果不準確。雙柱法中首先通過離子交換柱將低聚麥芽糖中的各組分按照聚合度實現(xiàn)分離,再使用氨基鍵合柱分離同分異構體。雙柱法雖然得到的結果準確,但對于聚合度較高的同分異構體也不能實現(xiàn)有效的分離,并且同一個樣品需要進行2次分析,分析過程較為繁瑣[4]。除了高效液相色譜法,也有毛細管電泳法應用于低聚異麥芽糖分析的報道,毛細管電泳結合激光誘導檢測器(CE-LIF)可以實現(xiàn)糖的快速分析,此方法靈敏度高,但屬于間接分析法,樣品需采用熒光試劑進行衍生化,衍生過程較為繁瑣[5]。高效陰離子交換色譜-脈沖安培檢測法(HPAEC-PAD)近年來被廣泛地應用于糖類的分析,目前已有應用于分析低聚木糖[6]、低聚果糖的報道[7]及其它糖類的分析[8-9],但還未有用于低聚異麥芽糖分析的報道。采用此方法分析糖的原理是基于糖是弱電解質,當溶液的pH在11以上,它能夠部分或全部以陰離子形式存在,可以吸附在陰離子交換柱上,并被不同濃度的堿溶液洗脫,配合脈沖安培檢測器可實現(xiàn)糖類的梯度洗脫及高靈敏度檢測[10]。理論上只要被分析的糖存在電離度上的差異,就可以實現(xiàn)分離,因此這種方法非常適合糖的同分異構體間的分離。本文采用CarboPakTMPA10為分析柱,建立了低聚異麥芽糖的HPAEC-PAD定性及定量的分析方法,實現(xiàn)了低聚異麥芽糖中15種組分包括同分異構體之間的一次性分離。
1 材料與方法
1.1 儀器與試劑
ICS-5000型離子色譜,配備高壓泵、柱溫箱、安培檢測器(Au工作電極,pH-Ag/AgCl參比電極)Chromeleon工作站(美國Dionex)。CarboPakTMPA10色譜柱(250 mm×4 mm),CarboPakTMPA10保護柱(50 mm×4 mm)(美國Dionex)。Millipore純水儀(美國millipore)。
葡萄糖,果糖,麥芽糖,麥芽三糖,異麥芽糖,異麥芽三糖,異麥芽四糖,異麥芽五糖,異麥芽六糖,異麥芽七糖,潘糖,海藻糖,麥芽酮糖,曲二糖,黑曲霉糖和50%NaOH試劑,購于Sigma-Aldrich公司。醋酸鈉(ICS級)購于Fisher公司。
低聚異麥芽糖漿:配制32%(W/W)底物濃度的麥芽糊精,用稀H2SO4調節(jié)pH至5.0。加入大麥β-淀粉酶OPTIMALT BBA(Genencor)和轉苷酶TG-L500(Genencor),酶用量分別為0.2 kg/t(干物)和1 kg/t(干物)。將此反應體系放入60℃水浴中反應40 h。反應結束后,糖液在10000 r/min離心5 min,取上清液稀釋后用0.22 μm濾膜過濾,用于色譜分析。
1.2 溶液配制
1.2.1 標準溶液配制
稱取一定量的標準樣品配制成1 g/L的母液,分裝后于-20℃存儲。臨用前將母液稀釋至所需濃度,得到不同濃度的標準樣品溶液。
1.2.2 淋洗液
250 mmol/L NaOH溶液:稱取20 g 50%NaOH溶液用純水定容至1 L,輕輕搖勻后立即轉入淋洗瓶中通入N2保護(5~8psi)。
500 mmol/L NaOH溶液:稱取40 g 50%NaOH溶液用純水定容至1 L,輕輕搖勻后立即轉入淋洗瓶中通入N2保護(5~8psi)。
200 mmol/L NaAc溶液:稱取16.4 g NaAc溶解,定容至1 L,搖勻后立即轉入淋洗瓶中通入N2保護(5~8psi)。
1.2.3 色譜分析條件
淋洗液A:純水;淋洗液B:250 mmol/L NaOH溶液;淋洗液C:200 mmol/L醋酸鈉溶液;淋洗液D:500 mmol/L NaOH溶液。梯度:0~12 min,22.5~32.5 mmol/L NaOH;12~13 min,32.5~21 mmol/L NaOH;13~31 min,21~90 mmol/L NaOH;31~36 min,90 mmol/L NaOH,0~4 mmol/L NaAc;36~39 min,90 mmol/L NaOH,4 mmol/L NaAc;39~48 min,90 mmol/L NaOH,4~100
mmol/L NaAc;48~52 min,90 mmol/L NaOH,100 mmol/L NaAc;52.1~56 min,200 mmol/L NaOH,4 mmol/L NaAc,56.1~65 min,22.5 mmol/L NaOH。進樣方式:手動進樣;柱溫:30℃;流速:1 mL/min。安培檢測器波形:carbohydrate standard quart。
2 結果與討論
2.1 分析條件的優(yōu)化
低聚異麥芽糖通常是以淀粉為底物經(jīng)過液化、糖化和轉苷得到,在轉苷反應中常使用具有轉糖苷作用的α-葡萄糖苷酶。在分析條件的優(yōu)化過程中發(fā)現(xiàn),除了常規(guī)的低聚異麥芽糖組分外,還檢測到了海藻糖、麥芽酮糖、曲二糖及黑曲霉糖等其它少量轉苷成分的存在。在傳統(tǒng)的檢測方中,這些少量的轉苷成分由于結構相近,不能實現(xiàn)有效分離。而在HPAECPAD的分析過程中只要這些組分在堿性條件下存在電離度的差異,就可以通過調節(jié)洗脫液的組成及濃度實現(xiàn)分離。NaOH作為常規(guī)洗脫液之一,在糖的分析中扮演著雙重角色,既為糖的電離提供堿性環(huán)境,同時也是洗脫劑,因此改變NaOH的濃度對于糖的保留時間有較大的影響[11-12],而NaAc相對于NaOH具有更強的洗脫力。在改變洗脫梯度的過程中發(fā)現(xiàn),對于異麥芽糖和麥芽酮糖的分離,提高NaAC濃度對于二者的影響遠大于NaOH濃度的改變,因此要實現(xiàn)二者的基線分離,可將洗脫液中NaAc的濃度降為0,同時提高NaOH的濃度。大幅增加NaOH濃度,可以提高聚合度較高糖的電離能力,增加其與色譜柱的結合能力,改善不同組分之間的分離。通過調節(jié)NaOH和NaAc的梯度,實現(xiàn)了低聚異麥芽糖中各組分的分離。在優(yōu)化了的分析條件下得到了低聚異麥芽糖漿及其標準樣品的譜圖,見圖1。

圖1 低聚異麥芽糖色譜圖
與HPLC的單柱法和雙柱法相比,HPAEC-PAD法可以一次性的實現(xiàn)低聚異麥芽糖中15種組分,包括5種二糖異構體,3種三糖異構體,及聚合度達到7的低聚糖的分離。與CE-LIF法相比可以實現(xiàn)非還原糖(海藻糖)及含有果糖基的二糖(麥芽酮糖)組分分離檢測[13]。
2.2 標準曲線和方法檢測限
將混合標準樣品母液稀釋6個不同的濃度,將這6個不同濃度的混合標準液采用已優(yōu)化的色譜條件進行分離,以濃度為縱坐標,峰面積為橫坐標,得到低聚異麥芽糖中各組分的標準曲線。低聚異麥芽糖各組分在表1所示的濃度內具有線性關系,線性相關系數(shù)均在0.9972以上。將低濃度樣品逐漸稀釋進樣直至相應的峰信號為空白樣品信噪比的3倍(S/N=3),得到此方法中各組分的檢測限(LOD),按照3倍檢測限確定各組分的定量限(LOQ)。方法的定量限及檢測限與HPLC法相比,實現(xiàn)了低聚異麥芽糖的10-6級的高靈敏度檢測。
2.3 實際樣品分析與精密度測定
采用本文所建立的方法對糊精轉苷得到的低聚異麥芽糖進行了測定,并加入2個濃度的各組分進行加標回收實驗,結果見表2。加標回收率為82.02%~116.37%,證實此方法具有較高的準確性。加標回收樣品測定的標準偏差為0.3%~4.78%。
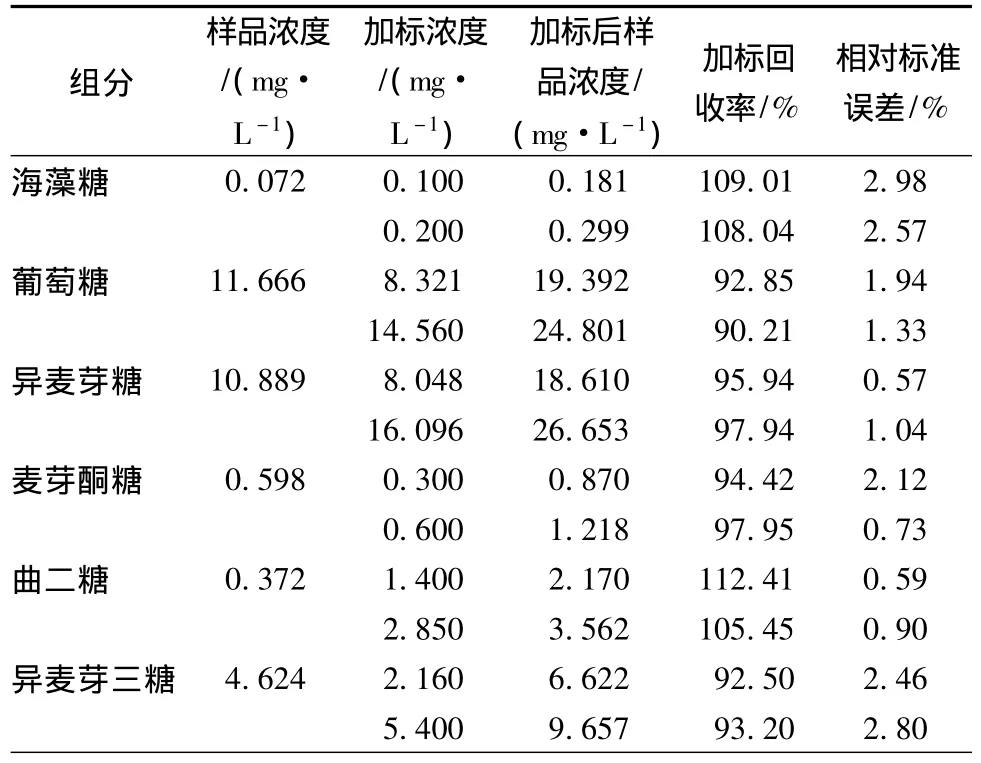
表2 低聚異麥芽糖各組分加標回收率(n=3)
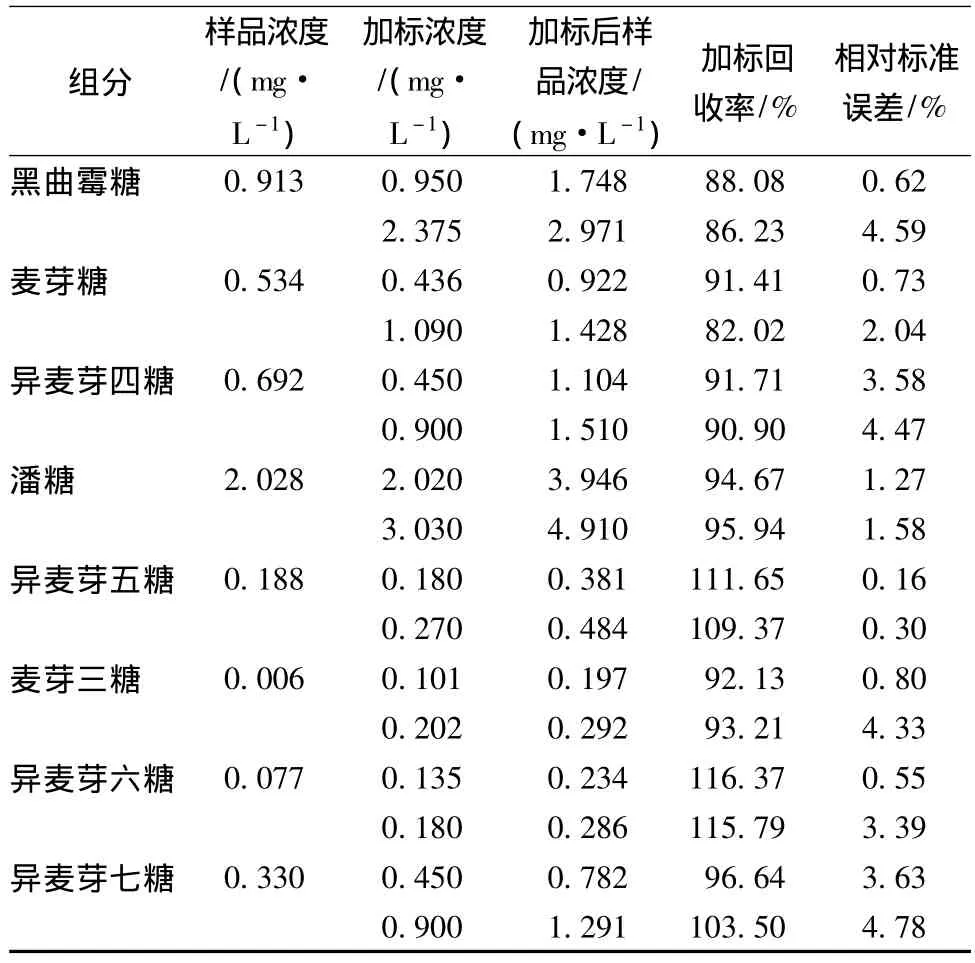
續(xù)表2
3 結論
采用CarboPakTMPA10色譜柱,建立了低聚異麥芽糖的HPAEC-PAD的分析方法。采用此方法實現(xiàn)了低聚異麥芽糖中15種組分,包括5種二糖異構體,3種三糖異構體,及聚合度達到7的低聚糖的一次性分離。通過定量及加標等實驗實現(xiàn)了低聚異麥芽糖的定量檢測并確定了方法的準確性。低聚異麥芽糖是我國功能性糖產(chǎn)業(yè)中非常重要的一種,雖然HPLC法是非常經(jīng)典的分析低聚異麥芽糖的方法,但HPAEC-PAD法可以更加深入的了解酶在低聚異麥芽糖生產(chǎn)中的作用方式及產(chǎn)品組分,為產(chǎn)品結構的優(yōu)化提供分析依據(jù)。
[1]尤新.功能性低聚糖生產(chǎn)與應用[M].北京:中國輕工業(yè)出版社,2004:1-3.
[2]Luca F A,Dolores M,Aranzazu G,et al.Transformation of maltose into prebiotic isomaltooligosaccharides by a novel&-glucosidase from Xantophyllomyces dendrorhous[J].Process Biochemistry,2007,42:1530-1536.
[3]鄭建仙.功能性低聚糖[M].北京:化學工業(yè)出版社,2004:2-6.
[4]Duan G,Li F,Shetty J K.US patent 7638151B2,2009.
[5]操麗麗,何進,林雁飛,等.低聚異麥芽糖產(chǎn)品中主要組分的毛細管電泳分析方法[J].食品與發(fā)酵工業(yè),2007,30(4):109-111.
[6]范麗,徐勇,連之娜,等.高效陰離子交換色譜-脈沖安培檢測法定量測定低聚木糖樣品中的低聚木糖[J].色譜,2011,29(1):75-78.
[7]Max F,Jinadevi S,Audrey A.Determination of complex polysaccharides by HPAE-PAD in foods:Validation using accuracy profile[J].Journal of Chromatography B,2009,877:2388-2395.
[8]Lee Y C.Carbohydrate analyses with high-performance anion-exchange chromatography[J].Journal of Chromatography A,1996,720:137-149.
[9]Greya C,Edebrinka P,Krooka M.Development of a high performance anion exchange chromatography analysis for mapping of oligosaccharides[J].Journal of Chromatography B,2009,877:1827-1832.
[10]牟世芬,劉克納.離子色譜方法與應用[M].北京:化學工業(yè)出版社.
[11]Cataldia T,Campa C,Angelotti M.Isocratic separations of closely-related mono-and disaccharides by high-performance anion-exchange chromatography with pulsed amperometric detection using dilute alkaline spiked with barium acetate[J].Journal of Chromatography A,1999,855:539-550.
[12]Kerherv P,Charrière B,Gadel F.Determination of marine monosaccharides by high-pH anion-exchange chromatography with pulsed amperometric detection[J].Journal of Chromatography A,1995,718:283-289.
[13]Bui A,Kocsis B,Kilar F.Methodology to label mixed carbohydrate components by APTS[J].Journal of Biochemistry and Biophysical Methods,2008,70:1313-1316.
ABSTRACTA one-step method for quantitatively determination of isomaltooligosaccharides(IMO)was developed using high performance anion exchange chromatography coupled with pulsed amperometric detection(HPAEC-PAD).The method was built on a CarboPakTMPA10 column using NaOH and NaAC as eluents.Using this method,besides conventional components of isomaltose,isomaltotriose,panose and some saccharides with higher DP were identified from IMO syrup,other transglycosylation saccharides such as trehalose,kojibiose,nigerose and maltulose were also detected from the syrup.Calibration was carried out by dissolving 15 kinds of standard samples containing glucose,fructose,maltose,maltotriose,isomaltose,isomaltotriose,isomaltotetraose,isomaltopentaose,isomaltohexaose,isomaltoheptaose,panose,trehalose,maltulose,kojibiose,and nigerose into a mixed solution.The standard solution was diluted to a calibration range from 0.032 to 25.975 mg/L.The calibration curves showed good linearity of IMO within this range.The detection limits(LODs)and the quatification limits(LQD)were 0.008~0.022 mg/L and 0.027~0.073 mg/L respectively,and the relative standard deviations were 82.02%~116.37%.This method was good and sensitive in the quantitative analysis of IMO.
Key wordsIMO,isomer,separation,HPAEC-PAD
Determination of Isomaltooligosaccharides by High Performance Anion Exchange Chromatography Coupled with Pulsed Amperometric Detection(HPAEC-PAD)
Zhang Xiao-ping,Duan Gang
(Genencor(China)Bio-products Co.,Ltd.,Wuxi 214028,China)
博士(段鋼為通訊作者)。
2012-03-20
猜你喜歡
現(xiàn)代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業(yè)技術(2016年15期)2016-12-01 05:31:22
當代經(jīng)濟研究(2016年5期)2016-12-01 03:12:05
現(xiàn)代農(nóng)業(yè)(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
中國中醫(yī)藥現(xiàn)代遠程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學學報(社會科學版)(2014年3期)2014-04-16 04:38:31
終身教育研究(2014年5期)2014-02-28 01:23:06
