IL-32γ對大鼠血管平滑肌細胞增殖與細胞周期的影響*
2012-09-14 06:46:20劉宇宏王莎莎沈凌汛徐玉蘭
中國病理生理雜志 2012年2期
關鍵詞:檢測
劉宇宏, 王莎莎, 沈凌汛, 徐玉蘭
(華中科技大學同濟醫學院附屬協和醫院風濕免疫科,湖北武漢430022)
類風濕關節炎(rheumatoid arthritis,RA)的早發心血管事件是同年齡、同性別未患該類疾病普通人群的4~5倍,其原因目前認為與RA慢性炎癥導致血管內膜損傷及中膜平滑肌細胞向內膜遷移并異常增生,從而加速動脈粥樣硬化的發生、演變與進展[1-2]。白細胞介素-32γ(interleukin-32γ,IL-32γ)是新近發現的一種致炎細胞因子[3-4],有研究發現它在RA的啟動與進展中發揮作用[5-7],但它對在RA動脈粥樣硬化進展中起關鍵作用的血管平滑肌細胞(vascular smooth muscle cells,VSMCs)增生是否產生影響尚不清楚。本研究擬通過觀察IL-32γ對體外培養的大鼠VSMCs增殖及細胞周期的影響,探討其在RA早發動脈粥樣硬化中的作用及機制。
材料和方法
1 材料
清潔級SD大鼠,4周齡,(180±20)g,雌雄不限,購自同濟醫學院實驗動物中心;含1×105U/L青霉素和100 mg/L硫酸鏈霉素的完全達氏修正依氏培 養 基 (Dulbecco's modified Eagle's medium,DMEM)、10%胎牛血清購自HyClone;II型膠原酶、二甲基亞砜(dimethyl sulfoxide,DMSO)、四甲基偶氮唑藍[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide,MTT]和吡咯烷二硫代氨基甲酸(pyrrolidine dithiocarbamate,PDTC)購自 Sigma-Aldrich;Annexin V-FITC/PI試劑盒購自南京博泰生物技術有限公司;鼠抗β-actin、組蛋白H3(histone H3)、核因子-κB p65(nuclear factor kappa B p65,NF-κB p65)、cyclin D1和增殖細胞核抗原(proliferating cell nuclear antigen,PCNA)抗體購自Santa Cruz;IL-32γ購自R&D。
2 方法
2.1 細胞培養 取4周齡SD大鼠胸主動脈,按常規組織貼塊方法分離、原代培養VSMCs,0.5%胰酶消化傳代,培養液為含10%胎牛血清的DMEM培養基,實驗采用3~8代細胞。
2.2 細胞增殖分析 取對數生長期的VSMCs,以含10% 胎牛血清的培養基制備為1×107/L細胞懸液,每孔200 μL分別接種于96孔培養板中培養,細胞同期化后分組處理。(1)對照組:IL-32γ的終濃度為0 μg/L;(2)實驗組:每孔分別加入 10、20、50 μg/L IL-32γ;(3)IL-32γ+PDTC 組:PDTC(終濃度20 μmol/L)預處理1 h后加入 IL-32γ(終濃度50 μg/L)。每組設4個復孔,然后在含10% 胎牛血清的培養液中分別培養24和48 h。干預結束時每孔加入MTT溶液(5 g/L)20 μL,繼續孵育4 h,棄培養液,再加入DMSO 200 μL,振蕩溶解細胞,用酶標儀檢測A490處的吸光度值。實驗重復3次。
2.3 細胞周期分析 VSMCs按2×108/L接種于24孔培養板中,細胞同期化后分組處理。實驗組加入IL-32γ 50 μg/L,對照組和 IL-32γ +PDTC 組處理方法如前述。每組設4個復孔,然后在含10% 胎牛血清的DMEM中培養24 h。收集細胞,用預冷的70%乙醇于4℃固定30 min,RNaseA(100 mg/L)37℃水浴30 min,加碘化丙啶(50 mg/L),以流式細胞儀(FACS Calibur)進行細胞周期分析。每個樣本檢測1×107個細胞。
2.4 免疫印跡法檢測cyclin D1和核內NF-κB p65蛋白的表達 按上述方法處理VSMCs,培養24 h后收集細胞1×109/L,提取胞質和核蛋白。取25 μg以10%SDS-PAGE電泳分離,于低溫100 V 3 h將蛋白轉至硝酸纖維素膜上,加封閉液2 h阻斷非特異性抗體結合位點,將膜與溶于封閉液中的Ⅰ抗室溫下孵育2 h,磷酸鹽緩沖液(phosphate-buffered saline,PBS)洗3次×5 min,加入辣根過氧化物酶標記的Ⅱ抗,室溫下結合1 h,PBS緩沖液沖洗后,標準ECL法底物發光并顯影。
2.5 免疫細胞化學染色法檢測PCNA的表達 6孔培養板中加人蓋玻片,VSMCs按1×109/L細胞密度接種,細胞長滿蓋玻片后按上述步驟加藥、培養。24 h和48 h收集蓋玻片,PBS液沖洗,純丙酮固定8 min,制成細胞爬片。I抗選用小鼠抗人PCNA單克隆抗體。按SP法染色試劑盒說明書操作步驟進行操作。每張細胞爬片隨機選取5個高倍視野,共計數1 000個細胞,計算陽性表達的細胞數占細胞總數的百分比。
3 統計學處理
結 果
1 VSMCs形態特點
將大鼠VSMCs分離、培養,倒置顯微鏡下觀察,細胞為梭形,有較長突起,呈典型“峰-谷”樣生長,見圖1。
2 IL-32γ對細胞增殖的影響
MTT比色法檢測表明,實驗組在接種24和48 h后的細胞數量與對照組比較顯著增多,且呈濃度和時間依賴性;提前1 h加入PDTC預處理,顯著抑制了IL-32γ的促增殖作用(分別 P<0.05,P<0.01),見表 1。
3 IL-32γ對細胞周期的影響
IL-32γ刺激細胞24 h后,實驗組處于G1期的細胞比例為(38±4)%,較對照組[(69±5)%]顯著降低,處于S+G2期的細胞比例為(62±5)%,較對照組[(31±3)%]明顯提高(均 P<0.01);加用PDTC預處理后,IL-32γ+PDTC組G1期的細胞比例為(60±5)%,處于S/G2期的細胞比例為(37±4)%,與實驗組比較差異顯著(均P<0.01),但與對照組比較差異無統計學意義,見圖2。

Figure 1.Morphology of VSMCs at the third passage under inverted microscope(×100).圖1 倒置顯微鏡下第3代VSMCs形態
表1 各組VSMCs的MTT檢測結果Table 1.Absorbance(A)values of VSMCs in different groups detected by MTT assay(±s.n=3)
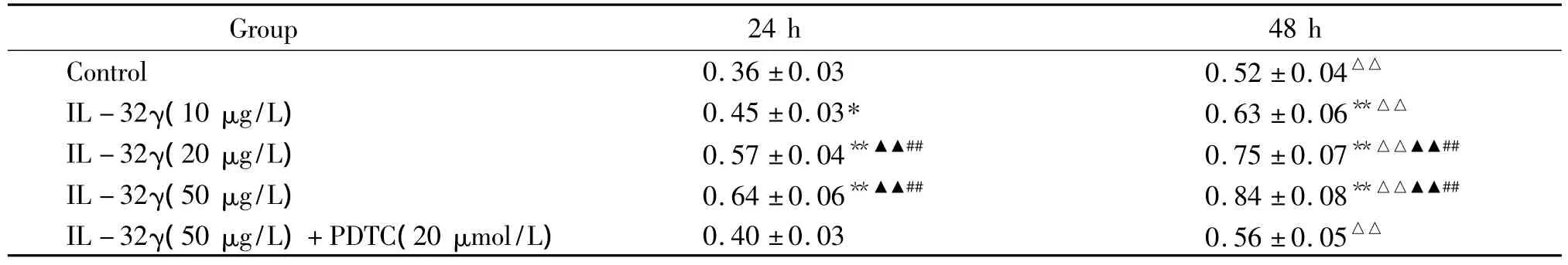
表1 各組VSMCs的MTT檢測結果Table 1.Absorbance(A)values of VSMCs in different groups detected by MTT assay(±s.n=3)
*P <0.05,**P <0.01 vs control at the same time;△△P <0.01 vs 24 h in the same group;▲▲P <0.01 vs IL-32γ(10 μg/L)at the same time;##P <0.01 vs IL-32γ (50 μmol/L)+PDTC(20 μmol/L)at the same time.
Group 24 h 48 h Control 0.36 ±0.03 0.52 ±0.04△△IL-32γ(10 μg/L) 0.45 ±0.03* 0.63 ±0.06**△△IL-32γ(20 μg/L) 0.57 ±0.04**▲▲## 0.75 ±0.07**△△▲▲##IL-32γ(50 μg/L) 0.64 ±0.06**▲▲## 0.84 ±0.08**△△▲▲##IL-32γ(50 μg/L)+PDTC(20 μmol/L) 0.40 ±0.03 0.56 ±0.05△△

Figure 2.Flow cytometry analysis of cell cycle圖2 流式細胞儀檢測細胞周期
4 IL-32γ對cyclin D1和NF-κB p65表達的影響
免疫印跡檢測結果顯示,實驗組cyclin D1和NF-κB p65的蛋白表達(分別為0.86±0.05和1.16±0.06)顯著高于對照組(分別為0.47±0.03和0.58±0.05)(均P<0.01);IL-32γ+PDTC組的 cyclin D1和NF-κB p65蛋白表達(分別為0.58±0.04和0.70±0.05)較實驗組顯著降低(均P<0.01),但仍高于對照組(均P<0.05),見圖3。
5 IL-32γ對PCNA表達的影響
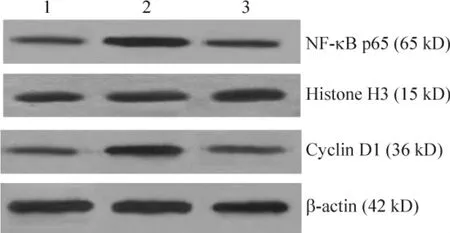
Figure 3.The expression levels of cyclin D1 and NF-κB p65 proteins in different groups(Western blotting).1:control group;2:IL-32γ group;3:IL-32γ+PDTC group.圖3 免疫印跡檢測各組細胞cyclin D1和NF-κB p65的蛋白表達
PCNA以細胞核棕黃色為陽性。實驗組第24和48 h的PCNA陽性表達量分別為(60.3±5.0)%和(73.1±6.2)%,顯著高于對照組的(38.1±4.2)%和(51.7±4.5)%(均 P<0.01);IL-32γ+PDTC組第24、48 h的PCNA陽性表達量分別為(40.1±4.0)%和(55.1±5.0)%,較實驗組顯著降低(均P<0.01),但與對照組比較差異無統計學意義。各組細胞形態未見明顯改變,見圖4。
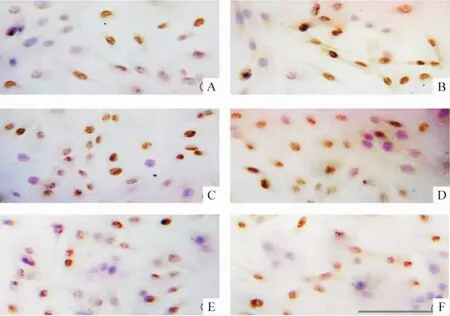
Figure 4.The immunocytochemical staining results of PCNA expression in each group(×200).A:control 24 h;B:control 48 h;C:IL-32γ 24 h;D:IL-32γ 48 h;E:IL-32γ+PDTC 24 h;F:IL-32γ+PDTC 48 h.圖4 免疫細胞化學染色法檢測各組細胞PCNA表達
討 論
類風濕關節炎患者的心血管發病率與死亡率明顯升高與其早發動脈粥樣硬化密切相關。近年研究發現,RA和動脈粥樣硬化的發病機制有著十分相似的特征,這些特征包括均有細胞因子參與、自體反應T細胞浸潤、黏附分子表達、膠原降解和新生血管形成等[1]。導致RA患者滑膜損傷的炎癥機制同時轉入了血管壁損傷并啟動了動脈粥樣硬化進程[1]。
VSMCs的異常增殖是動脈粥樣硬化過程中的一個關鍵因素。作為一個病理過程,其異常增殖受多方面的因素的影響與調控,諸如細胞因子[8]、趨化因子、生長因子等。晚近有研究發現IL-32γ高表達于RA患者的血管壁,且與內皮功能失調密切相關[9],但它對VSMCs增殖的影響尚不清楚。本研究結果顯示,與對照組相比,10 μg/L以上的 IL-32γ可以濃度和時間依賴性地使VSMCs增殖加快。提示IL-32γ對體外培養的VSMCs有促增殖作用。PCNA含量隨細胞不同周期而改變,其量的變化與DNA合成一致,可反映細胞增殖活性的變化。免疫細胞化學結果顯示,PCNA在IL-32γ組中的表達水平明顯高于對照組,表明細胞的DNA合成增多,即細胞增殖加快,與MTT檢測法的結果一致。這與文獻報道IL-32表達上調可以促進造血干細胞增殖[10]、敲除IL-32基因可以抑制胰腺癌細胞增殖是一致的[11],表明IL-32γ的確是一個與細胞增殖相關的細胞因子。
細胞周期長短是影響細胞增殖快慢的重要因素之一。本研究結果發現,當用50 μg/L IL-32γ刺激48 h后,實驗組處于G1期的細胞比例較對照組降低,處于S+G2期的細胞比例較對照組提高。表明IL-32γ的促細胞增殖作用與加速細胞周期轉化密切相關。
細胞周期受細胞周期素(cyclins)的調控。Cyclin D1在調控細胞通過G1期檢驗點發揮重要作用,通過G1期檢驗點的細胞將不可逆轉地完成整個細胞分裂過程[12]。Cyclin D1表達上調可以加速細胞通過整個細胞周期[13-15]。因此,我們進一步檢測IL-32γ對cyclin D1表達的影響。結果表明,cyclinD1在實驗組中的表達顯著高于對照組。表明IL-32γ在cyclin D1表達的調控中發揮重要的作用。上調cyclin D1表達可能是IL-32γ加速細胞周期轉化的原因之一。
已知NF-κB可以通過轉錄調節cyclin D1而控制細胞生長[16]。有研究報道IL-32γ可激活人樹突狀細胞、單核細胞和小鼠巨噬細胞的NF-κB信號通路[17-18],但在VSMCs中是否存在這種機制尚未見報道。本實驗發現,實驗組核內NF-κB p65的表達顯著高于對照組。提示IL-32γ可誘導RA-FLS中NF-κB活化。為了進一步了解該信號通路的活化與IL-32γ促細胞增殖作用間的相關性,我們較IL-32γ提前1 h加用NF-κB抑制劑PDTC預處理VSMCs,結果顯示,PDTC顯著抑制IL-32γ的促細胞增殖反應、促細胞周期轉化和上調NF-κB p65與cyclin D1表達的作用。提示IL-32γ的確能通過激活NF-κB信號通路上調cyclin D1表達,進而縮短細胞周期和促進細胞增殖。
總之,本實驗發現,IL-32γ能夠在體外促進VSMCs增殖。表明IL-32γ作為炎癥級聯反應過程中的一個重要細胞因子,不僅參與了RA關節損傷的啟動與進展,而且在RA早發動脈粥樣硬化中發揮重要作用,可能是RA及其心血管并發癥治療的一個新靶標。
[1] Hürlimann D,Forster A,Noll G,et al.Anti-tumor necrosis factor-alpha treatment improves endothelial function in patients with rheumatoid arthritis[J].Circulation,2002,106(17):2184-2187.
[2] Full LE,Monaco C.Targeting inflammation as a therapeutic strategy in accelerated atherosclerosis in rheumatoid arthritis[J].Cardiovasc Ther,2011,29(4):231-242.
[3] Choi JD,Bae SY,Hong JW,et al.Identification of the most active interleukin-32 isoform[J].Immunology,2009,126(4):535-542.
[4] Felaco P,Castellani ML,De Lutiis MA,et al.IL-32:a newly-discovered proinflammatory cytokine[J].J Biol Regul Homeost Agents,2009,23(3):141-147.
[5] Alsaleh G,Sparsa L,Chatelus E,et al.Innate immunity triggers IL-32 expression by fibroblast-like synoviocytes in rheumatoid arthritis[J].Arthritis Res Ther,2010,12(4):R135.
[6] Kim YG,Lee CK,Oh JS,et al.Effect of interleukin-32γ on differentiation of osteoclasts from CD14+monocytes[J].Arthritis Rheum,2010,62(2):515-523.
[7] Joosten LA,Netea MG,Kim SH,et al.IL-32,a proinflammatory cytokine in rheumatoid arthritis[J].Proc Natl Acad Sci U S A,2006,103(9):3298-3303.
[8] Kim HM,Bae SJ,Kim DW,et al.Inhibitory role of magnolol on proliferative capacity and matrix metalloproteinase-9 expression in TNF-α-induced vascular smooth muscle cells[J].Int Immunopharmacol,2007,7(8):1083-1091.
[9] Nold-Petry CA,Nold MF,Zepp JA,et al.IL-32-dependent effects of IL-1beta on endothelial cell functions[J].Proc Natl Acad Sci U S A,2009,106(10):3883-3888.
[10] Moldenhauer A,Futschik M,Lu H,et al.Interleukin 32 promotes hematopoietic progenitor expansion and attenuates bone marrow cytotoxicity[J].Eur J Immunol,2011,41(6):1774-1786.
[11] Nishida A,Andoh A,Inatomi O,et al.Interleukin-32 expression in the pancreas[J].J Biol Chem,2009,284(26):17868-17876.
[12] Pardee AB.A restriction point for control of normal animal cell proliferation[J].Proc Natl Acad Sci USA,1974,71(4):1286-1290.
[13] Hunter T,Pines J.Cyclins and cancer II:cyclin D and CDK inhibitors come of age[J].Cell,1994,79(4):573-582.
[14] 喬禮芬,徐永健,劉先勝,等.周期蛋白D1在蛋白激酶C調控哮喘大鼠氣道平滑肌細胞增殖中的作用[J].中國病理生理雜志,2008,24(11):2214-2219.
[15] Hinz M,Krappmann D,Eichten A,et al.NF-κB function in growth control:regulation of cyclin D1 expression and G0/G1-to-S-phase transition[J].Mol Cell Biol,1999,19(4):2690-2698.
[16] Guttridge DC,Albanese C,Reuther JY,et a1.NF-κB controls cell growth and differentiation through transcriptional regulation of cyclin D1[J].Mol Cell Biol,1999,19(8):5785-5799.
[17] Jung MY,Son MH,Kim SH,et al.IL-32γ induces the maturation of dendritic cells with Th1-and Th17-polarizing ability through enhanced IL-12 and IL-6 production[J].J Immunol,2011,186(12):6848-6859.
[18] Kim SH,Han SY,Azam T,et al.Interleukin-32:a cytokine and inducer of TNFα[J].Immunity,2005,22(1):131-142.
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
