柱前衍生化高效液相色譜法測定蓮子心中去甲烏藥堿含量
2012-10-25 00:49:28田海峰衣濤金東日
延邊大學學報(自然科學版) 2012年2期
田海峰, 衣濤, 金東日*
(1.延邊大學理學院 化學系,吉林 延吉133002;2.吉林煙草工業有限責任公司,吉林 延吉133001)
柱前衍生化高效液相色譜法測定蓮子心中去甲烏藥堿含量
田海峰1, 衣濤2, 金東日1*
(1.延邊大學理學院 化學系,吉林 延吉133002;2.吉林煙草工業有限責任公司,吉林 延吉133001)
以9-芴甲基-N-琥珀酰亞胺基碳酸酯(Fmoc-OSu)為熒光衍生化試劑,建立了柱前熒光衍生化反相高效液相色譜法分析去甲烏藥堿的高靈敏分析方法.在硼酸緩沖溶液(p H=8.5)中,去甲烏藥堿與Fmoc-OSu在溫和的反應條件下反應生成具有熒光性的去甲烏藥堿衍生物.采用UltimateR○XB-C18(5μm,150 mm×4.6 mm i.d.)色譜柱,用乙腈-水(體積分數比為85∶15)等溶液強度洗脫,以λ=265 nm為激發波長,λ=315 nm為發射波長,對去甲烏藥堿衍生物進行了檢測.實驗結果顯示:去甲烏藥堿在0.05~20μg/m L范圍內呈良好的線性關系(r=0.999 8),其最低檢出限(S/N=3)為5.0 ng/m L,加標回收率為94.1%~105.9%,RSD為2.74%(n=5).本方法靈敏度高、選擇性好、準確度高,能夠滿足含去甲烏藥堿中藥材的質量控制.
去甲烏藥堿;柱前熒光衍生化;HPLC;Fmoc-OSu;蓮子心
去甲烏藥堿(higenamine,HG)是烏頭、附子、蓮子心等中藥中所含有的強心的有效成分.藥理實驗表明,去甲烏藥堿對心血管系統有正力性和正時性效應[1],其特性類似于多巴胺丁胺,有望成為一種新的臨床心肌負荷藥物.近年來研究發現,去甲烏藥堿對iNOS m RNA的表達、脂多糖LPS誘導的NO產物以及對血小板聚集和血栓形成都具有抑制作用,而且對動物實驗性DIC也具有改善作用[2].目前,去甲烏藥堿的常用測定方法有HPLC-UV[3]、HPLC-ECD[4]、HPLC-ESI-MS[5]、HPLC-FLD[6]等,其中HPLC-UV方法檢測靈敏度較低,不適于含微量去甲烏藥堿試樣的分析;HPLC-ECD和HPLC-ESI-MS方法檢測靈敏度雖然較高,但由于ECD和MS檢測器較為昂貴,因此限制了其廣泛的應用.本文研究者曾用手性熒光衍生化試劑建立了分離去甲烏藥堿對映體的HPLC-FLD[7]方法.本文以9-芴甲基-N-琥珀酰亞胺基碳酸酯(Fmoc-OSu)為熒光衍生化試劑,用HPLC-FLD建立了去甲烏藥堿的測定方法,并用此方法測定了蓮子心中去甲烏藥堿的含量.
1 實驗部分
1.1 儀器與試劑
儀器有Shimadzu LC-10AVP高效液相色譜儀(日本島津公司),RF-10AXL熒光檢測器,Class-VP色譜工作站,AS20500A超聲波清洗器(天津奧特賽恩斯儀器有限公司),HB-100恒溫金屬浴(杭州大和熱電子有限公司).
去甲烏藥堿(純度大于97%)由江西中藥研究所提供,Fmoc-OSu(純度大于98%)購于J&K CHEMICAL LTD,乙腈、甲醇為色譜純(山東禹王實業有限公司),氫氧化鉀、氯化鉀為優級純,硼酸為分析純試劑.
配制1mg/m L的去甲烏藥堿和5 mmol/L的Fmoc-OSu,于冰箱中保存,實驗前稀釋到所需濃度;用0.2 mol/L氫氧化鉀溶液調節硼酸緩沖溶液p H值至8.5.
1.2 衍生化反應
分別取一定濃度的去甲烏藥堿標準溶液、Fmoc-OSu乙腈溶液(待測物濃度的125倍)和硼酸緩沖溶液(p H=8.5)各10μL混合于聚丙烯管中,用渦旋攪拌器攪拌1 min,在室溫下反應50 min.
1.3 樣品前處理
將蓮子心研磨成粉末,精確稱取蓮子心粉末2.50 g放入100 m L錐形瓶中,加入25 m L甲醇,在超聲波清洗器中超聲30 min(50℃下),過濾.上述操作重復進行3次,合并濾液.將濾液在45℃下減壓蒸餾濃縮旋至近干,用乙腈-水(體積分數比為1∶1)定容至25 m L.經0.45μm微孔濾膜過濾,取1 m L濾液用水定容至200 m L.
1.4 色譜條件
采 用UltimateR○XB-C18(5μm,150 mm×4.6 mm i.d.)色譜柱,流動相為乙腈-水(體積分數比為85∶15),柱溫為室溫,進樣量為10μL;熒光檢測波長為λex=265 nm,λem=315 nm.
2 結果與討論
2.1 衍生化反應條件
Fmoc-OSu作為一種柱前衍生化試劑,具有過量的衍生化試劑不干擾分離、其衍生物穩定性較好、基體干擾不明顯、衍生選擇性較好等[7]特點,已用于氨基酸、多肽的分離分析.因此,本研究以Fmoc-OSu作為分離分析去甲烏藥堿的衍生化試劑.圖1為去甲烏藥堿與Fmoc-OSu的衍生化反應式.
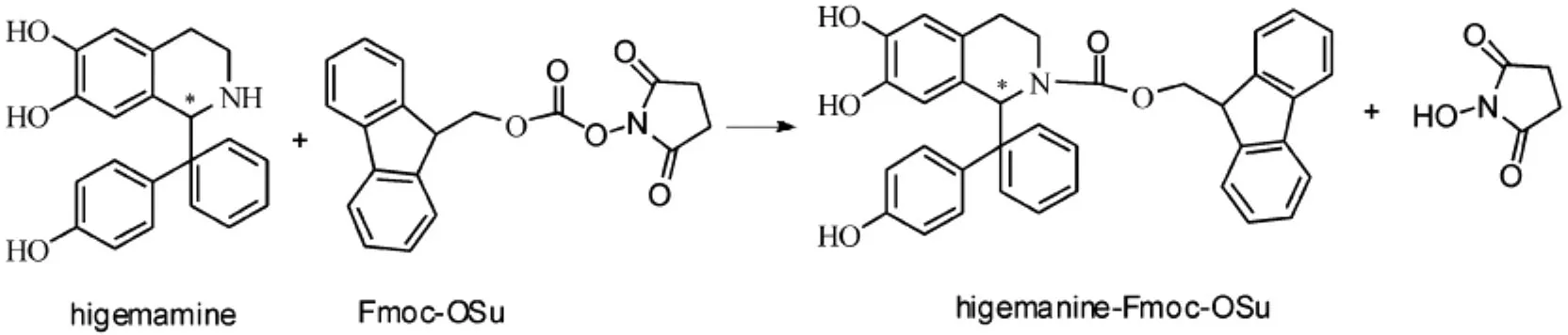
圖1 去甲烏藥堿與Fmoc-OSu的衍生化反應式
影響去甲烏藥堿與Fmoc-OSu衍生化反應的因素有反應介質的酸度、反應時間、衍生化試劑的濃度等.首先,根據文獻[7]以硼酸緩沖溶液為反應介質,探討緩沖溶液的p H值對衍生化效率的影響.從圖2可知,硼酸緩沖溶液的p H值為8.5時去甲烏藥堿衍生物的峰面積最大,所以本實驗將p H值為8.5的硼酸緩沖溶液作為反應介質.其次,在室溫條件下,觀察反應時間對衍生化反應的影響.圖3顯示,反應開始時去甲烏藥堿衍生物的峰面積隨反應時間的增加而逐漸增加,當反應時間達到50 min時,衍生物的峰面積最大且趨于恒定,因此衍生化反應時間選為50 min.最后,考察衍生化試劑濃度對衍生化反應的影響.實驗表明,對于濃度為0.04μmol/m L的去甲烏藥堿,Fmoc-OSu的濃度為5μmol/m L(衍生化試劑濃度/去甲烏藥堿濃度=125)時,衍生物的峰面積最大且趨于恒定,所以將衍生化試劑的比選定為125(Fmoc-OSu濃度/去甲烏藥堿濃度).

圖2 硼酸緩沖溶液p H值對衍生化反應的影響

圖3 反應時間對衍生化反應的影響
2.2 檢測波長及色譜條件
以λ=265 nm為激發波長,λ=315 nm為發射波長,對去甲烏藥堿衍生物進行檢測.采用UltimateR○XB-C18(5μm,150 mm×4.6 mm i.d.)色譜柱,進行衍生化反應液中去甲烏藥堿衍生物的分離.在甲醇-水、乙腈-水、磷酸鹽緩沖液(p H=3.0)-乙腈、磷酸二氫鈉(p H=3.0)-乙腈等洗脫液中,當以乙腈-水(體積分數比為85∶15)作為流動相,流速為1 m L/min時,去甲烏藥堿衍生物與基質中的其他峰得到了很好的分離(見圖4).根據與空白和樣品色譜圖的比較,確定保留時間為13.6 min的峰為去甲烏藥堿衍生物,其他峰為過量Fmoc-OSu和反應副產物.

圖4 去甲烏藥堿與過量Fmoc-OSu衍生化反應后的色譜圖:a為Fmoc-OSu(空白),b為HG-Fmoc-OSu衍生物
2.3 線性關系和檢出限
去甲烏藥堿在0.05~20μg/m L范圍內呈良好的線性關系 (r=0.999 8),其回歸線性方程為Y=1.181 76×106X+18 320.427 25(Y為去甲烏藥堿衍生物峰面積,X 為去甲烏藥堿濃度).當S/N=3時,檢出限為5.0 ng/m L,比HPLCUV[3]方法高10倍.
2.4 精密度和穩定性
將濃度為10μg/m L的去甲烏藥堿標準溶液衍生化后,同1天連續進樣5次,置于冰箱(4℃)中的衍生化產物連續5 d進樣.結果顯示,日內精密度和日間精密度分別為2.74%和9.14%.
2.5 加標回收率
采用標準加入法對分析方法進行準確度驗證.取1.25 g蓮子心粉末6份,分別加入1.2 mg去甲烏藥堿標樣,考察樣品的加標回收率.結果顯示,樣品的回收率為88.3%~112.5%,相對標準偏差為5.8%~8.4%(n=5).
2.6 蓮子心中去甲烏藥堿含量的測定
利用本方法測定蓮子心中的去甲烏藥堿含量.蓮子心提取液的衍生化色譜圖見圖5.在色譜圖中,實驗保留時間為13.69 min的峰是去甲烏藥堿衍生物,其含量測定結果為94.0 mg/100 g.
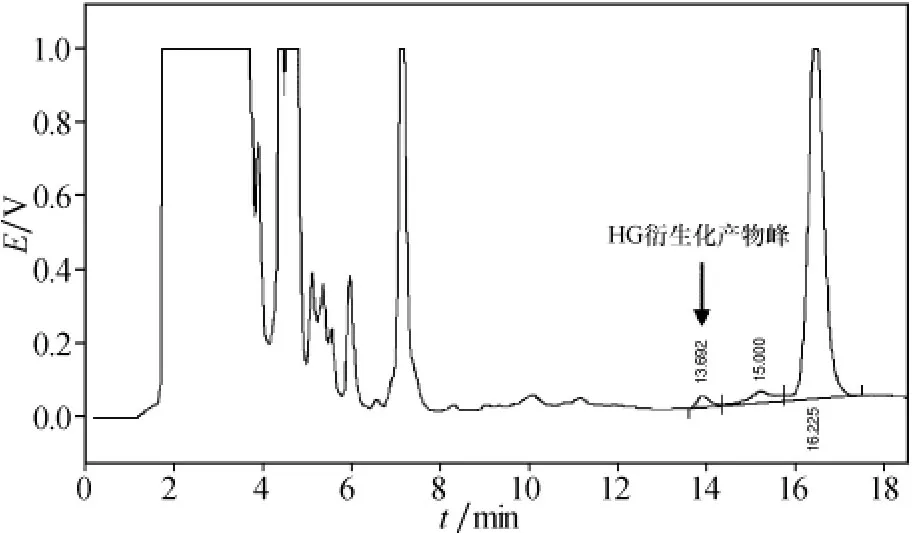
圖5 蓮子心提取液的衍生化色譜圖
3 結論
研究了HPLC-FLD分離去甲烏藥堿的Fmoc-OSu柱前衍生化條件和色譜條件.使用該方法可使衍生化反應定量地進行,其反應條件溫和,衍生化產物較穩定,而且過量的衍生化試劑和副產物均不干擾待測物的分離和檢測.該方法選擇性好,檢測靈敏度高,可用于含去甲烏藥堿的中藥材和相關制劑的質量控制.
[1] Kosuge T,Yokota M.Studies on cardiac principle of aconite root[J].Chem Pharm Bull(Tokyo),1976,24:176-178.
[2] 周素娟,杜貴友.去甲烏藥堿對心血管系統的影響[J].中國中藥雜志,2003,28(10):910-913.
[3] 陳寶玲,鄭英麗,陸直,等.HPLC法測定去甲烏藥堿的含量[J].中國新藥雜志,2004,13(7):628-630.
[4] Lo C F,Chen C M.Determination of higenamine in plasma and urine by high performance liquid chromatography with electrochemical detection[J].J Chromatogr B,1994,655:33-39.
[5] Feng S,Hu P,Jiang J.Determination of higenamine in human plasma and urine using liquid chromatography coupled to positive electrospray ionization tandem mass pectrometry[J].J Chromatogr B,2011,879:763-768.
[6] 丁雅韻,謝孟峽.Fmoc-OSu作為液相色譜分離分析氨基酸的柱前衍生試劑研究[J].分析實驗室,2001,20(Suppl):138-139.
[7] Hong H,Lee Y I,Jin D.Determination of R-(+)-higenamine enantiomer in Nelumbo nucifera by highperformance liquid chromatography with a fluorescent chiral tagging reagent[J].Microchemcal J,2010,96:374-379.
Determination of higenamine in Nelumbo nucifera by HPLC with precolumn derivatization
TIAN Hai-feng1, YI Tao2, JIN Dong-ri1*
(1.Department of Chemistry,College of Science,Yanbian University,Yanji 133002,China;2.JILin Tobacco Industrial Co.,LTD,Yanji 133001,China)
A sensitive method for determination of higenamine based on a derivatization reaction with a fluorescent tagging reagent,9-fluorenylmethoxycarbonylsuccinimide(Fmoc-OSu)is developed.The Fmoc-OSu preferably reacts with higenamine under mild reaction conditions in the presence of borate buffer(p H=8.5)to produce the corresponding fluorescent derivative with an excitation maximum at 265 nm and an emission maximum at 315 nm.The derivative of higenamine are efficiently resolved on a UltimateR○XB-C18(5μm,150 mm×4.6 mm i.d.)column by an isocratic elution with water-acetonitrile(85∶15)mobile phase.The linear range is 0.05-20μg/m L(r=0.999 8)for higenamine.The average recovery of the higenamine is 94.1%-105.9%(RSD 2.74%).The limits of detection(S/N=3)per injection is 5.0 ng/m L.The developed method is applied successfully to the determination of higenamine in embryo of Nelumbo nucifera,a Chinese herbal medicine.
higenamine;precolumn derivatization method;HPLC;Fmoc-OSu;Nelumbo nucifera
O657.72
A
1004-4353(2012)02-0150-04
2012-01-20
*通信作者:金東日(1965—),男,教授,研究方向為藥物分析.
