RP-HPLC法測定蒼耳子中蒼術苷
2012-11-01 14:08:00劉玉紅王明奎易進海
中成藥 2012年11期
劉玉紅,朵 睿,2,黃 蛟,王明奎,易進海*
(1.四川省中醫藥科學院,四川 成都 610041;2.瀘州醫學院,四川 瀘州,646000;3.中國科學院成都生物所,四川 成都 610041)
蒼耳子是菊科植物蒼耳Xanthium sibiricum Patr.的干燥成熟帶總苞的果實,具有散風寒、通鼻竅、祛風濕之功效,用于風寒頭痛、鼻塞流涕、鼻鼽、鼻淵等癥,是鼻淵之要藥[1],為臨床常用有毒中藥。《本草品匯精要》記載蒼耳子“有毒”。《南方主要有毒植物》記載: “蒼耳,有毒部位,全株;以果實為最毒”。因蒼耳服用過量或炮制不當引起的中毒甚至死亡的病例時有發生[2-3],病理特征表現為動物和人在誤食一定量后出現血糖下降,昏迷,嚴重者出現腎衰竭甚至死亡;慢性中毒多因初服時未出現明顯的不良反應而長期服用,結果導致蓄積中毒,引起心肌及肝功能損害[4]。現代化學和毒理研究表明蒼耳子的毒性成分主要為水溶性苷類成分:蒼術苷(atractyloside)與羧基蒼術苷(carboxyatractyloside)以及其它苷類衍生物等[5]。蒼術苷及羧基蒼術苷可抑制體內ADP/ATP對蛋白的轉運,并帶來嚴重的血糖下降,從而導致體內代謝紊亂[6-8],與蒼耳子中毒的表現一致,因此被認為是蒼耳子的主要毒性成分。建立蒼耳子中蒼術苷的測定方法,對蒼耳子的毒性成分進行監控,保證臨床用藥安全是十分必要的。蒼耳子中蒼術苷的測定尚未見文獻報道,其它樣品中蒼術苷的測定方法文獻報道有酶聯免疫法[9]、TLC斑點測試[10]、GC-MS[11]、HPLC-ELSD[12]、LC-API-MS[13]等,這些方法在定量的準確性或方法的普適性方面存在一定的局限。本實驗首次建立了RP-HPLC測定蒼耳子中蒼術苷的方法,該方法簡便、快速,重復性好,為控制蒼耳子的質量和毒性提供了參考。
1 材料
Agilent 1200型高效液相色譜儀 (包括四元泵,DAD檢測器,柱溫箱,自動進樣器,工作站);KQ—100超聲波清洗儀 (昆山市超聲儀器有限公司),Mettler Toledo XS205型電子天平 (瑞士),Millipore Milli-Q超純水系統 (美國)。
實驗樣品購于四川、遼寧、山東、河北、內蒙古、新疆、甘肅等地共35批蒼耳子藥材和飲片,由四川省中醫藥科學院舒光明研究員鑒定。蒼術苷對照品自制,純度為98.1%,經理化性質和光譜數據分析,鑒定其結構。乙腈為美國Fisher色譜純;水為Millipore Milli-Q超純水系統制得的超純水;其余試劑均為分析純。
2 方法與結果
2.1 色譜條件 色譜柱為Agilent ZORBAX SB-phenyl(4.6 mm×250 mm,5 μm)柱;流動相為乙腈-0.01 mol/L磷酸二氫鈉溶液 (20∶80);體積流量1.0 mL/min;檢測波長203 nm;柱溫35℃。對照品溶液及供試品溶液的色譜圖見圖1。同時對樣品中蒼術苷的200~400 nm紫外光譜圖與對照品的紫外光譜圖進行比較,表明樣品峰無其它雜質干擾。
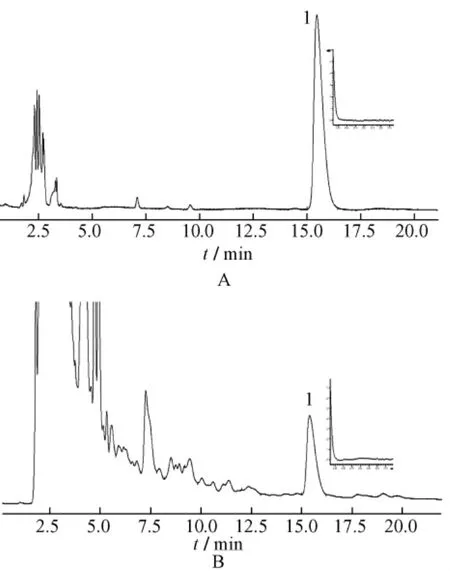
圖1 蒼術苷對照品 (A)及蒼耳子供試品 (B)的HPLC圖Fig.1 HPLC chromatograms of atractyloside(A)and Xanthii Fructus(B)
2.2 對照品溶液的制備 精密稱取蒼術苷對照品11.29 mg,置10 mL量瓶中,加10%甲醇溶液溶解并稀釋至刻度,搖勻。再精密量取上述對照品溶液1 mL,置10 mL量瓶中,加10%甲醇溶液稀釋至刻度,搖勻,制成每1 mL中含蒼術苷0.1129 mg的對照品溶液。
2.3 供試品溶液的制備 取蒼耳子粉未 (過三號篩)1 g,精密稱定,置具塞錐形瓶中,加水20 mL,精密稱定質量,超聲 (功率250 W,頻率40 kHz)處理40 min,取出,放冷,再稱定質量,用水補足減失質量,離心 (12000 r/min,5 min),取上清液作為供試品溶液。
2.4 方法學考察
2.4.1 線性關系 精密吸取蒼術苷對照品各1、3、6、9、12、15、20 μL,注入液相色譜儀中,以進樣量 (μg)為橫坐標,對照品峰面積 (A)為縱坐標,繪制標準曲線。結果蒼術苷的標準曲線為Y=458.9X-3.884(r=0.9999),線性范圍為0.1129 ~2.258 μg。
2.4.2 定量限和檢出限 將2.2項下的溶液,用10%甲醇溶液稀釋成0.01129 mg/mL的溶液,進樣測定,按10倍和3倍信噪比計算出蒼術苷的定量限和檢出限分別約為34 ng、11.3 ng。
2.4.3 精密度試驗 精密吸取同一供試品溶液(樣品01)10 μL,注入液相色譜儀,連續進樣5次,按2.1項下色譜條件測定,蒼術苷的峰面積的RSD為0.58%,表明儀器精密度好。
2.4.4 穩定性試驗 精密吸取同一供試品溶液(樣品01)10 μL,注入液相色譜儀,分別于0、1、4、8、24 h檢測,記錄色譜圖,蒼術苷的峰面積的RSD為0.83%,表明供試品溶液于24 h內穩定。
2.4.5 重復性試驗 取同一蒼耳子 (樣品01)粉未,精密稱取約1 g,共5份,按2.3項下方法制備供試品溶液,精密吸取供試品溶液10 μL,分別注入液相色譜儀,記錄色譜圖,計算出蒼術苷質量分數為2.14 mg/g、RSD為1.0%,表明該方法重復性好。
2.4.6 加樣回收試驗 取已知蒼術苷含有量的樣品 (樣品01,2.14 mg/g),0.5 g,共6份,精密稱定,準確加入適量對照品,照2.3項下操作,測定,計算回收率。結果見表1。
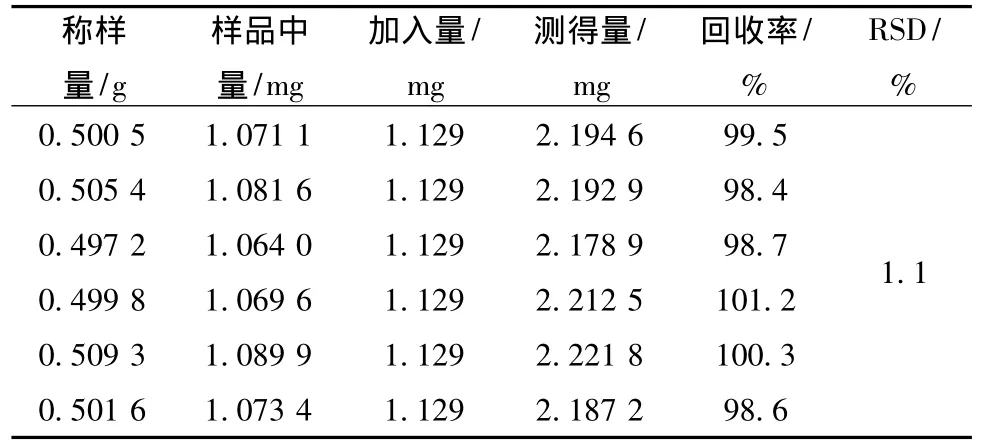
表1 蒼術苷的加樣回收率實驗 (n=6)Tab.1 Recovery of atractyloside
2.5 樣品測定 照2.3項下方法制備35批次蒼耳子樣品的供試品溶液,分別精密吸取10~20 μL注入液相色譜儀測定,在2.1項色譜條件下進行測定,計算各樣品中蒼術苷,結果見表2。
3 討論
3.1 提取溶劑和樣品制備方法的考察 蒼術苷為水溶性苷類。本實驗比較了水、25%甲醇、50%甲醇對蒼術苷超聲提取的效果,3種溶劑測定同一樣品的含有量分別為0.214%、0.191%,0.0723%,表明不同溶劑提取對蒼術苷有顯著影響,以水提取效果最佳。又以水為提取溶劑,比較了超聲提取30、40、60 min的差異,結果表明提取 40 min即可。
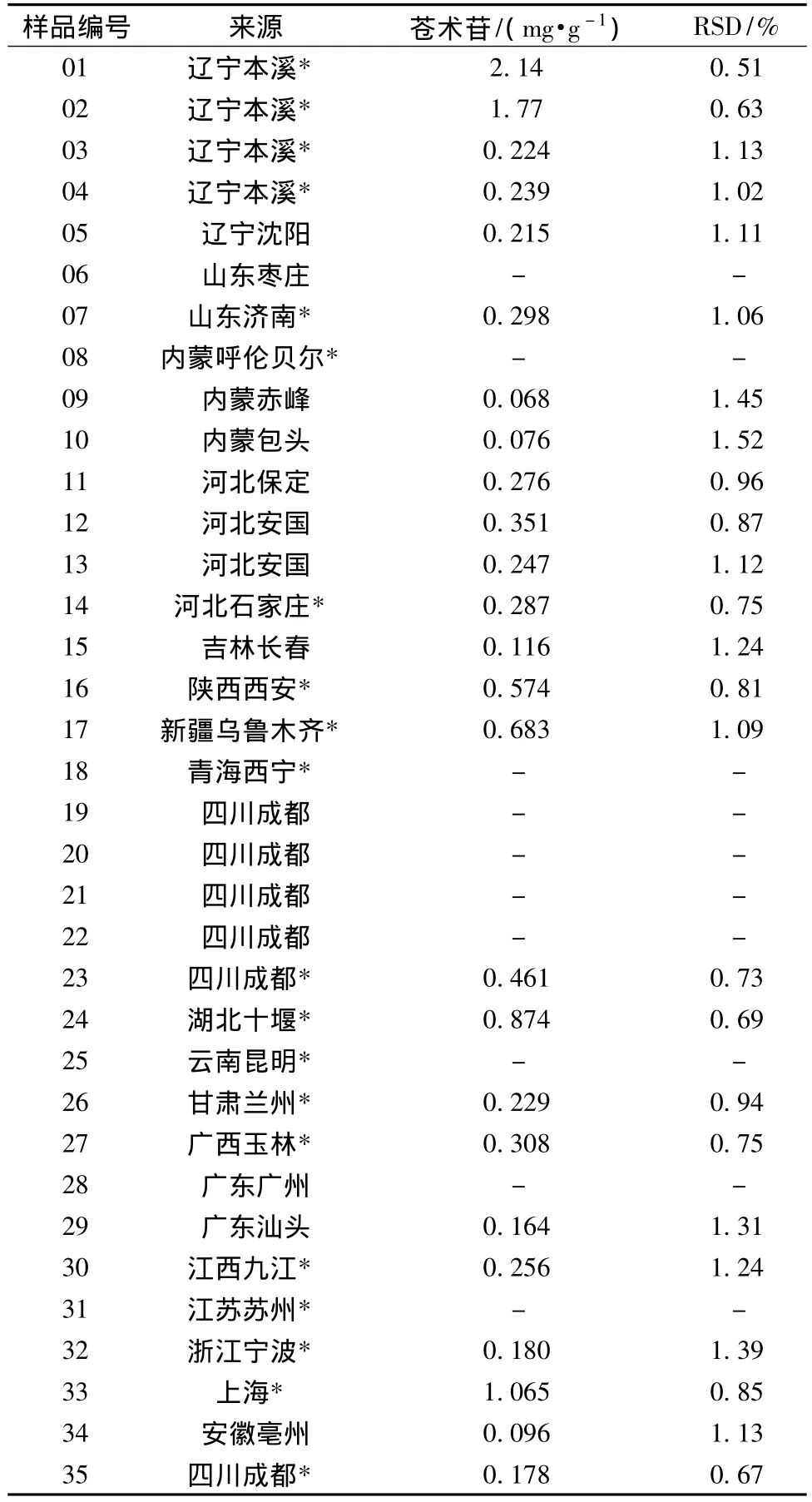
表2 蒼耳子樣品中蒼術苷的測定結果 (n=3)Tab.2 Contents of atractyloside in Xanthii Fructus
3.2 色譜條件的選擇和優化
3.2.1 流動相系統的篩選 參照文獻[13],采用甲醇-醋酸鹽緩沖液系統,結果基線噪音太大,該系統不適合紫外檢測器短波長檢測。又自行摸索比較了甲醇-磷酸水、乙腈-磷酸水、乙腈-磷酸鹽等系統,結果以乙腈-磷酸鹽系統較好,但仍存在色譜峰拖尾嚴重的問題,故對不同色譜柱的分離效果進行比較。
3.2.2 色譜柱的篩選 比較了Phenomenex LUNA C18(2)(4.6 mm ×250 mm,5 μm),Agilent Eplise XDB-C18柱 (4.6 mm ×250 mm,5 μm),親水性C18柱 Phenomenex Gemini C18110A(4.6 mm×250 mm,5 μm),苯基柱 Agilent ZORBAX SB-phenyl(4.6 mm×250 mm,5 μm)的分離效果,結果以苯基柱分離效果最佳,峰形較為對稱。
3.2.3 測定波長的選擇 蒼術苷為貝殼杉烯苷類化合物,DAD掃描可見僅有末端吸收,故采用203 nm為測定波長,結果表明靈敏度較高,待測成分基線分離,重現性良好,符合定量測定的要求。
3.3 測定結果 35批次蒼耳子藥材及飲片的測定結果,表明不同來源蒼耳子中蒼術苷的含有量差異很大,有必要制定蒼術苷的限量標準。有報道稱南方產蒼耳子低毒,北方產蒼耳子毒性較大,本實驗測定結果與此結論有相同趨勢。
[1]馬 萍,李 紅.蒼耳子的研究進展[J].中草藥,1999,30(8):634.
[2]張學梅,張重華.蒼耳子中毒及毒性研究進展[J].中西醫結合學報,2003,51(1):71.
[3]閻芳華.過量服用蒼耳子中毒致死一例[J].山西醫藥雜志,2007,36(3):212.
[4]梁曉燕,朱會友,王建麗,等.小劑量蒼耳子中毒1例[J].實用中醫藥雜志,1998,14(10):45.
[5]汪 洋.中藥蒼耳子的毒性物質基礎及中毒機制研究[D].上海:第二軍醫大學,2010
[6]Obatomi D K,Bach P H.Biochemistry and toxicology of the diterpenoid glycoside atractyloside[J]. Food Chem Toxicol,1998,36(4):335-346.
[7]Klingenberg M.The ADP,ATP shuttle of the mitochondrion[J].Trends Biochem Sci,1979,4(11):249-252.
[8]阮貴華,李攻科.蒼耳子的化學成分及其分離分析研究進展[J].中成藥,2008,30(3):421.
[9]Bye S N,Coetzer T H T,Dutton M F.An enzyme immunoassay for atractyloside,the nephrotoxin of Callilepis laureola(Impila)[J].Toxicon,1990,28(8):997-1000.
[10]Steenkamp V,Stewart M J,Zuckerman M.Detection of poisoning by Impila(Callilepis laureola)in a mother and child[J].Human ExpToxicol,1999,18(10):594-597.
[11]Laurens J B,Bekker L C,Steenkamp V,et al.Gas chromatographic-mass spectrometric confirmation of atractyloside in a patient poisoned with Callilepis laureola[J].J Chromatogr B,2001,765(2):127-133.
[12]Stafford C G,St Claire R L 3rd.High-performance liquid chromatographic analysis of the lactone and carboxylate forms of a topoisomerase I inhibitor(the antitumor drug G1147211)in plasma[J].J Chromatogr B Biomed Appl,1995,663(1):119-126.
[13]Steenkamp P A,Harding N M,van Heerdeen F R,et al.Determination of atractyloside in Callilepis laureola using solid-phase extraction and liquid chromatography-atmospheric pressure ionization-mass spectrometry[J].J Chromatogr A,2004,1058(1/2):153-162.
