多肽抑制劑抑制淀粉質多肽42構象轉換的分子動力學模擬和結合自由能計算
2012-12-11 09:06:06董曉燕都文婕劉夫鋒
物理化學學報 2012年11期
關鍵詞:結構
董曉燕 都文婕 劉夫鋒
(天津大學化工學院生物工程系,天津300072;天津大學,系統生物工程教育部重點實驗室,天津300072)
多肽抑制劑抑制淀粉質多肽42構象轉換的分子動力學模擬和結合自由能計算
董曉燕 都文婕 劉夫鋒*
(天津大學化工學院生物工程系,天津300072;天津大學,系統生物工程教育部重點實驗室,天津300072)
應用分子動力學模擬和結合自由能計算方法研究了多肽抑制劑KLVFF、VVIA和LPFFD抑制淀粉質多肽42(Aβ42)構象轉換的分子機理.結果表明,三種多肽抑制劑均能夠有效抑制Aβ42的二級結構由α-螺旋向β-折疊的構象轉換.另外,多肽抑制劑降低了Aβ42分子內的疏水相互作用,減少了多肽分子內遠距離的接觸,有效抑制了Aβ42的疏水塌縮,從而起到穩定其初始構象的作用.這些抑制劑與Aβ42之間的疏水和靜電相互作用(包括氫鍵)均有利于它們抑制Aβ42的構象轉換.此外,抑制劑中的帶電氨基酸殘基可以增強其和Aβ42之間的靜電相互作用(包括氫鍵),并降低抑制劑之間的聚集,從而大大增強對Aβ42構象轉換的抑制能力.但脯氨酸的引入會破壞多肽的線性結構,從而大大降低其與Aβ42之間的作用力.上述分子模擬的結果揭示了多肽抑制劑KLVFF、VVIA和LPFFD抑制Aβ42構象轉換的分子機理,對于進一步合理設計Aβ的高效短肽抑制劑具有非常重要的理論指導意義.
分子動力學模擬;阿爾茨海默病;淀粉質多肽;多肽抑制劑;構象轉換
1 引言
阿爾茨海默病(AD)是一種以進行性記憶和認知功能損傷為特征的退行性神經系統疾病,目前確切病因尚不清楚.臨床表現為認知和記憶功能不斷惡化,日常生活能力進行性減退,并伴有各種神經癥狀和行為障礙.1,2AD是老年人的多發病和常見病,尤其是隨著世界人口老齡化的加劇,已成為人類的第四號殺手,將對社會和經濟發展造成巨大的負面影響.
目前AD的病理變化存在多種假說,如Aβ假說、膽堿能損害假說和炎癥反應假說等.3,4由于AD的主要病理特征為腦神經細胞外出現β-淀粉樣多肽(Aβ)聚集形成的老年斑和腦神經細胞內Tau蛋白異常聚集形成的神經纖維纏結等,因此Aβ假說得到了深入的發展.越來越多的證據表明,Aβ是引起AD的原發性病理因子.5Aβ假說認為,Aβ的錯誤折疊和聚集形成以β-折疊為主的聚集體,會引發一系列復雜的反應,包括突觸變化和炎癥反應等,最終出現神經元功能失調、斑塊形成、神經原纖維纏結等病理現象.6
Aβ是淀粉質前體蛋白的水解產物,通常為含有39-43個氨基酸殘基的多肽,以Aβ40和Aβ42為主,其中又以Aβ42的聚集沉淀趨勢最強.7研究表明,Aβ分子首先從初始的α-螺旋轉變成以β-折疊為主的二級結構,然后相互聚集形成寡聚體和纖維.8-10早期研究認為,只有Aβ聚集形成的淀粉質斑或纖維才具有神經毒性,而近來越來越多的研究證明,可溶性Aβ寡聚體和原細纖維體的神經毒性更大.11但無論是Aβ寡聚體還是成熟纖維(或者是兩者),只要阻止Aβ的構象轉變或聚集就可從根本上消除Aβ所引發的神經毒性.因此,目前對于AD的治療策略主要著眼于Aβ聚集抑制劑的開發.
現有的Aβ聚集抑制劑主要有:有機小分子、蛋白質和多肽.12-15由于多肽類抑制劑具有較好的生物相容性、毒副作用小、分子量小、易于透過血腦屏障等優點,因此近年來備受關注.16-18目前研究開發的多肽類抑制劑大多為Aβ的片段(KLVFF和VVIA)或其衍生物(LPFFD).19-21大量研究表明,這些多肽類抑制劑可以與Aβ發生結合從而阻止Aβ聚集和毒性的產生.22-26但由于Aβ構象轉換迅速,用目前的實驗技術無法檢測,導致多肽類抑制劑抑制Aβ構象轉換的作用機理無法用現有的實驗方法進行研究.然而分子動力學模擬的出現彌補了現有實驗研究的不足,已廣泛應用于Aβ構象轉換和聚集及其抑制的研究中.27例如,Xu等10利用全原子分子動力學模擬探明了Aβ40從初始的α-螺旋到β-折疊構象轉換的分子機理,并發現四個甘氨酸對于上述的構象轉換起到非常重要的作用,將其突變成丙氨酸后就能有效地抑制其構象轉換.Wei等28利用復制交換分子動力學模擬解析了Aβ25-35聚集成二聚體和纖維的結構形態.
此外,分子動力學模擬也常用來研究抑制劑抑制Aβ構象轉換和聚集的分子機理.作者前期利用分子動力學模擬解析了海藻糖抑制Aβ16-22聚集和茶多酚EGCG抑制Aβ42構象轉換的分子機理.9,29Yang等19以分子對接獲得的Aβ42-LPFFD復合物為初始結構,利用全原子分子動力學模擬解析了多肽抑制劑LPFFD抑制Aβ42構象轉換的分子機理.研究結果表明LPFFD通過破壞Aβ42的鹽橋D23-K28來抑制Aβ42的構象轉換.Viet等30利用分子動力學模擬和結合自由能計算方法解析了短肽抑制劑KLVFF和LPFFD影響Aβ16-22聚集和Aβ40構象轉換的分子機理,并解析了抑制劑與Aβ之間的親和力與其抑制能力之間的關系.但該研究沒有解析短肽抑制劑KLVFF和LPFFD抑制Aβ42構象轉換的分子機理.現有大量實驗研究表明,位于Aβ42 C端的多肽片段VVIA能夠有效抑制Aβ的聚集沉淀.31但至今還沒有利用分子動力學模擬解析位于Aβ42 C端的多肽VVIA抑制Aβ42構象轉換的分子機理.此外,從這三種多肽抑制劑的氨基酸序列可以看出,KLVFF和LPFFD除了含有疏水性氨基酸殘基外還含有帶電氨基酸殘基,而VVIA僅含有疏水氨基酸殘基.此外,LPFFD中脯氨酸殘基的引入對抑制Aβ42構象轉換的影響尚不清楚.因此,利用分子動力學模擬和結合自由能計算方法研究比較這三種多肽抑制劑抑制Aβ42的構象轉換以及它們和Aβ42之間的親和作用,進一步解析多肽抑制劑中帶電氨基酸和脯氨酸的引入對于短肽抑制劑抑制Aβ構象轉換的影響有助于理解短肽抑制劑抑制Aβ構象轉換和聚集之間的相互關系,對于多肽類AD抑制劑的合理設計具有重要的理論指導和實際意義.
本文以Aβ42為研究對象,采用全原子模型的分子動力學模擬和結合自由能計算方法,考察了常見的三種多肽類抑制劑KLVFF、VVIA和LPFFD對Aβ42構象變化的影響,通過分析在不同抑制劑存在下Aβ42的二級結構變化、Aβ42的側鏈接觸、抑制劑和Aβ42之間的接觸距離、抑制劑和Aβ42之間的結合自由能和氫鍵分析,來進一步解析三種多肽抑制劑抑制Aβ42構象轉變的作用機理.
2 模擬方法
2.1 模擬體系構建
Aβ42的初始結構從蛋白質數據庫(PDB)獲得, PDB號為1IYT.32Aβ42的初始結構含有兩個α-helix,分別定義為helix-1(8-25位殘基)和helix-2(28-39位殘基)(圖1A).其氨基酸序列為:DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVIA.
三種多肽類抑制劑KLVFF、VVIA和LPFFD的化學結構式如圖1(B,C,D)所示,它們均利用SYBYL軟件繪制得到.這些抑制劑的N末端用乙酰基修飾,C末端用亞氨基修飾.
首先將Aβ42放入一個立方體盒子(8 nm×8 nm× 8 nm)中,然后將多肽抑制劑放入該盒子,使其隨機分布,并與Aβ42之間的最小距離為0.5 nm.隨后將盒子中加滿水,并去除與多肽或抑制劑重疊的水分子;最后,用不同數目的Na+或Cl-替代同等數目的水分子使模擬體系保持電中性.其中,水溶液中添加3個Na+,抑制劑KLVFF溶液中添加5個Cl-, VVIA溶液中添加3個Na+,LPFFD溶液中添加11個Na+.采用三次能量最小化優化該體系:首先,固定Aβ42和抑制劑分子的結構和位置不變,僅讓水分子的結構和位置發生變化;其次,僅固定Aβ42的結構和位置,使抑制劑和水分子的結構和位置發生變化;最后,使體系內所有分子都可以自由運動,利用能量最小化來優化上述模擬體系.
2.2 分子動力學模擬
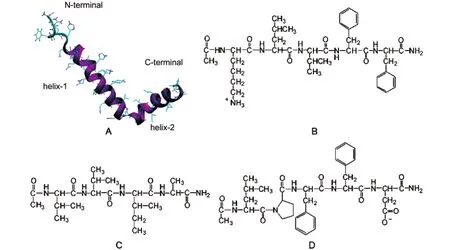
圖1 Aβ42的初始結構(A)與三種多肽抑制劑KLVFF(B),VVIA(C)和LPFFD(D)的化學結構Fig.1 Initial structure ofAβ42(A)and chemical structures of the three peptide inhibitors KLVFF(B),VVIA(C),and LPFFD(D)The main chain ofAβ42 is shown by a NewCartoon model and its side chains are represented by a line.
將上述優化好的體系進行分子動力學模擬,每個體系模擬時間為80 ns.各模擬體系的詳細信息,如表1所示.模擬過程中,為了使模擬結果更具有代表性,對相同抑制劑條件下的同一體系通過改變動力學模擬參數中的隨機數,使體系中各原子獲得不同的初始速度,從而得到三條不同的模擬軌跡.對于圖2-7,除了圖2,3和5之外,文中的所有分析都基于三次模擬結果的平均.圖2和圖3是利用GROMACS軟件自帶的程序基于一條模擬軌跡計算獲得.圖5中Aβ42和多肽抑制劑之間相互作用的典型構象利用VMD1.9.1軟件制作.
表1 模擬體系的參數Table 1 Parameters of the simulation systems
本文采用GROMACS 4.0.5分子動力學模擬軟件,33選擇GROMOS 96力場,34水分子采用單點電荷(SPC)模型.35利用Lenard-Jones函數計算范德華作用力,非鍵截斷距離設定為1.4 nm,非鍵作用原子列表每5個步長更新一次;利用particle-mesh Ewald方法計算靜電相互作用.36采用Verlet leapfrog算法37對每一步的運動方程進行求解,經過積分得到新時刻各原子的坐標,積分步長為2 fs.同時,采用LINCS算法38對所有成鍵原子之間的相對距離進行固定,以減小計算量.所有模擬均在等溫等壓(NPT)系綜中進行,溫度300 K,通過V-rescale方法控制溫度,時間常數設為0.1 ps.壓力為1.013×105Pa,壓力控制采用Berendsen方法,39壓力耦合常數為0.5 ps.所有分子動力學模擬計算均在曙光TC2600刀片服務器上完成.
2.3 數據分析方法
2.3.1 Aβ42的二級結構分析
采用GROMACS軟件中的附屬程序do_dssp計算Aβ42的二級結構,考察多肽的二級結構隨模擬時間的變化.40
2.3.2 Aβ42側鏈接觸圖計算
采用GROMACS軟件中的附屬程序g_mdmat計算多肽側鏈接觸圖.截斷距離設為0.9 nm.
2.3.3 抑制劑和Aβ42之間接觸數和接觸距離的計算
抑制劑和Aβ42之間的接觸數和接觸距離均采用GROMACS附屬程序g_mindist進行計算.其中,計算抑制劑和Aβ42之間的接觸數時所用的截斷距離為0.6 nm.
2.3.4 結合自由能計算
抑制劑和Aβ42之間的結合自由能(ΔGbind)采用分子力學-泊松-玻爾茲曼-溶劑可及表面積(MMPBSA)方法進行計算分析.41,42計算公式如下:

其中,ΔEMM為分子內能,通過分子力學方法計算獲得,ΔGsol為溶劑化自由能,ΔS為熵.分子內能主要包括范德華作用能(ΔEvdw)和靜電相互作用能(ΔEelec).而溶劑化自由能主要包含極性溶劑化自由能(ΔGPB)和非極性溶劑化自由能(ΔGnps).極性溶劑化自由能利用泊松-玻爾茲曼方程計算獲得,溶質和溶劑的介電常數分別為1和80.非極性溶劑化自由能由溶劑空穴作用項和溶質-溶劑范德華作用項組成,如公式(2)所示.

其中,γ和b分別為2.27 kJ·mol-1·nm-2和3.85 kJ· mol-1.SASA為溶劑可及表面積.
為了詳細分析抑制劑和Aβ42之間的結合自由能,作者將抑制劑和Aβ42之間的自由能分解為極性作用能ΔGPBelec和非極性作用能ΔGnonpolar.在接下來的分析中,ΔGPBelec主要包含靜電相互作用和極性溶劑化自由能,因此可將ΔGPBelec認為是抑制劑和Aβ42之間的靜電作用貢獻.而ΔGnonpolar主要包含范德華作用(ΔEvdw)和非極性溶劑化自由能,因此可以將其作為抑制劑和Aβ42之間的疏水作用貢獻.
2.3.5 氫鍵分析
本文中抑制劑和Aβ42之間的氫鍵作用利用GROMACS附屬程序g_hbond進行分析.當供體原子D與受體原子A之間的距離小于0.35 nm,且D―H―A之間的夾角大于120°時,即認為供體原子D和受體原子A之間形成氫鍵.最理想的氫鍵的鍵角是D―H―A為180°.
3 結果與討論
3.1 Aβ42的二級結構分析
圖2為Aβ42在水和不同多肽抑制劑溶液中的二級結構隨模擬時間的變化.在模擬過程中,利用不同的顏色來表示多肽的二級結構.其中圖2A為Aβ42在水溶液條件下的二級結構變化.從該圖可以看出,在水溶液中隨著模擬的進行,Aβ42的N和C端的初始α-螺旋(α-helix)結構被破壞,而轉換成轉角(turn)、無規卷曲(coil)和β-折疊(β-sheet)等結構;但在抑制劑KLVFF存在情況下,雖然Aβ42 C端的25-42位殘基的初始α-螺旋被破壞并轉變成轉角,彎曲(bend)和無規卷曲等結構,并在之后的模擬過程中一直穩定存在;但其N端6-24位殘基的α-螺旋結構得到了很好的保持(圖2B所示).其主要原因是抑制劑上的帶正電荷的賴氨酸(K)能夠和6-24位殘基中的帶負電荷氨基酸(D7,E11,E23和D24)之間形成較強的靜電相互作用,從而維持了N端6-24位殘基的初始α-螺旋構象.圖2C為Aβ42在抑制劑VVIA存在情況下的二級結構變化.從該圖可看出,Aβ42 N端8-12位氨基酸從45 ns開始逐步轉化為富含5-螺旋(5-helix),無規卷曲和彎曲等構象,并在隨后的模擬中始終穩定存在;C端的構象在5 ns左右被破壞,出現少量的β-折疊和轉角結構,但穩定性較差, 40 ns開始逐步被轉角和彎曲等結構所替代;13-25位殘基在模擬過程中始終維持著初始的α-螺旋結構(圖2C所示).圖2D為抑制劑LPFFD抑制Aβ42二級結構變化的情況.從該圖可以看出,N末端的構象維持較好,30 ns左右8-13位殘基開始出現轉角結構,并一直穩定維持到80 ns;22-28位殘基的構象發生轉變,在35 ns以后出現較多的5-螺旋結構.此外,C端32-37位殘基在模擬的80 ns內均維持初始的α-螺旋結構,但C末端38-40位殘基在模擬初始時就出現轉角結構且在80 ns的模擬過程中一直穩定存在;且在模擬過程中沒有出現β-折疊二級結構.從圖2D可以看出,抑制劑LPFFD除了能夠抑制Aβ42 N端2-7位殘基的構象轉換,還能夠維持C端32-37位殘基的初始α-螺旋構象.其主要原因是抑制劑上的帶負電荷天冬氨酸(D)可以和5位精氨酸(R)和28位賴氨酸(K)之間形成較強的靜電相互作用,從而維持其相關區域的初始構象.上述模擬結果表明,三種多肽抑制劑都能夠抑制Aβ42 N端的構象轉換,維持Aβ42的初始α-螺旋構象,并抑制β-折疊結構的形成.此外,多肽抑制劑上的帶電氨基酸殘基對于抑制Aβ42的構象轉換和維持其初始構象具有非常重要的作用.
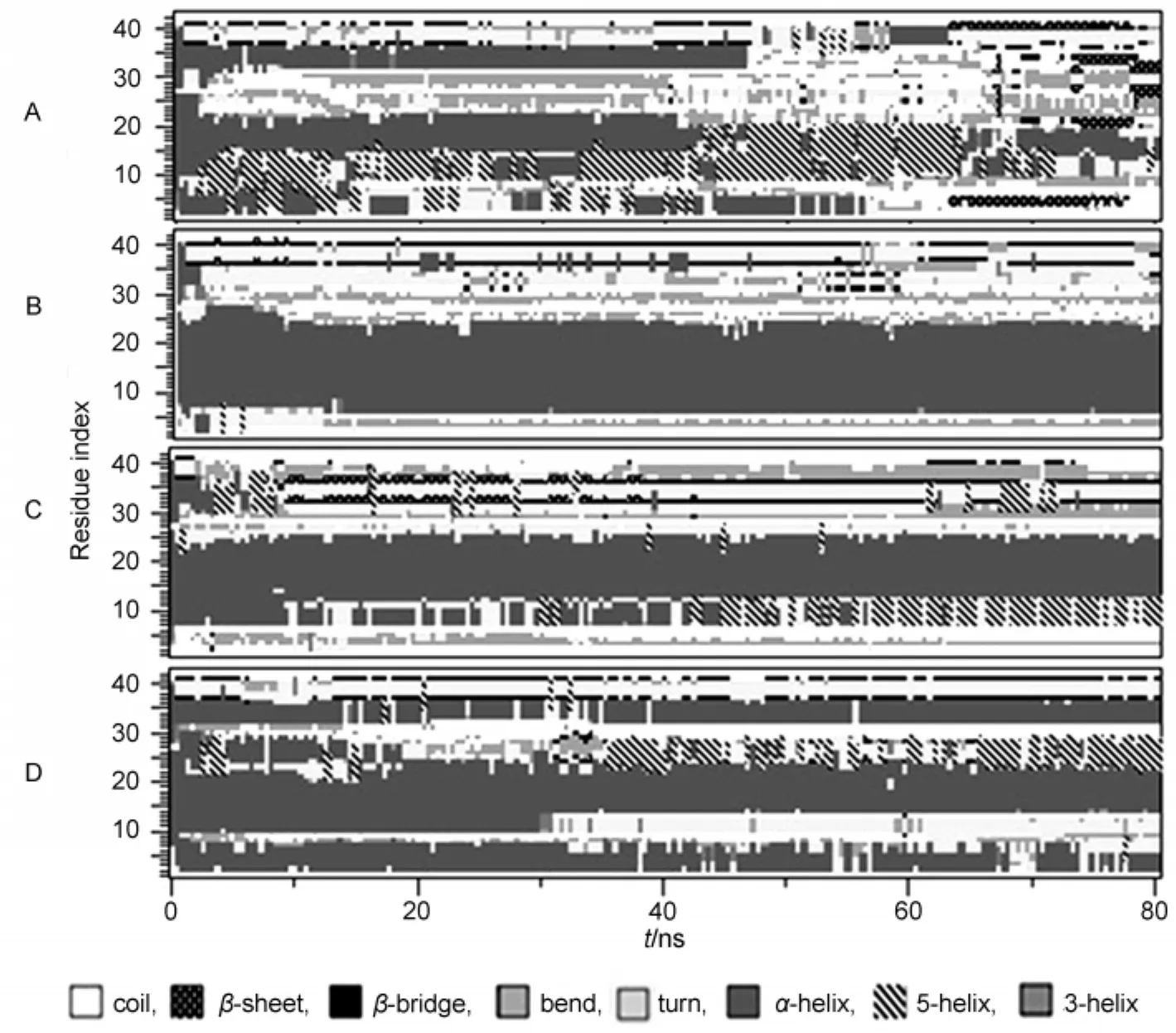
圖2 Aβ42的二級結構隨模擬時間的變化Fig.2 Secondary structures as a function of simulation time forAβ42(A)without inhibitor;(B)KLVFF;(C)VVIA;(D)LPFFD.Secondary structure is color-coded.
3.2 Aβ42側鏈接觸圖分析
多肽側鏈的接觸圖(contact map)常被用來表征多肽分子內疏水相互作用.43,44圖3為Aβ42在水溶液和不同抑制劑溶液中的側鏈接觸圖.Aβ42中殘基側鏈之間接觸距離的大小用圖中不同的顏色表示,其中白色和黑色分別表示Aβ42側鏈之間的距離為0和0.9 nm.顏色越接近于白色,表示相關殘基側鏈之間的距離越近;由于對角線表示的是相同殘基側鏈之間的接觸距離,其值為0,因此在圖中顯示白色.
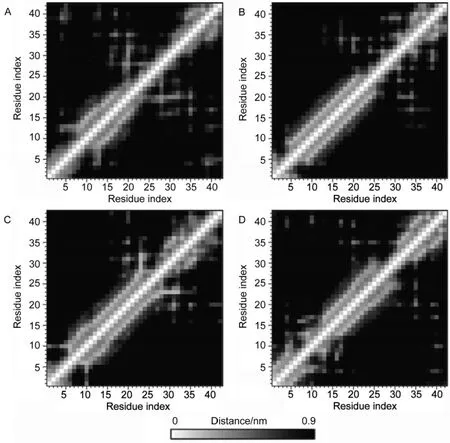
圖3 Aβ42的側鏈接觸圖Fig.3 Contact maps of the side chains ofAβ42(A)no inhibitor,(B)KLVFF,(C)VVIA,(D)LPFFD;Each square provides the mean average distance between any atoms of two residues ofAβ42.
可以看出,在水溶液中,Aβ42的遠距離側鏈之間的接觸數比較多,這主要是由于Aβ42在分子內疏水作用下發生塌縮使其N末端和C末端靠近的結果(圖3A),這與作者早期的研究結果44類似.而在三種多肽抑制劑存在的情況下Aβ42的遠距離側鏈的接觸數明顯減少,如圖3B,3C和3D所示.這證明了抑制劑的存在抑制了Aβ42的疏水塌縮,從而降低了N和C末端之間的接觸數.此外,圖3B中Aβ42的氨基酸側鏈之間的遠距離和近距離接觸數都比圖3C和3D少,說明KLVFF抑制劑對Aβ42疏水塌縮的抑制作用效果要好于另兩個抑制劑VVIA和LPFFD.由于Aβ42是疏水性很強的多肽,其分子內的疏水相互作用在其構象轉換和聚集過程中發揮重要的作用.因此,抑制Aβ42的疏水塌縮,即抑制Aβ42分子內的疏水相互作用,是抑制其構象轉換的前提.因此,多肽抑制劑的存在減少了Aβ42分子內遠距離的接觸,有效抑制了Aβ42的疏水塌縮,降低了Aβ42分子內的疏水相互作用,從而起到穩定其初始構象的作用.
3.3 多肽抑制劑和Aβ42之間的接觸數和接觸距離分析
為了進一步分析三種抑制劑抑制Aβ42構象轉換的能力,計算了三種抑制劑和Aβ42的接觸數和接觸距離.抑制劑和Aβ42之間的接觸數和接觸距離可以用于表征抑制劑和Aβ42之間的作用力大小.抑制劑與Aβ42之間的接觸數多和距離短,表明抑制劑和Aβ42之間的作用力強;反之,則說明抑制劑和Aβ42之間的作用力弱.圖4為Aβ42和抑制劑之間的接觸數隨模擬時間的變化.從圖4可以看出,抑制劑KLVFF和Aβ42之間的接觸數上升最快,在模擬初始的1 ns內就急劇上升到3300左右,并且隨著模擬的進行而緩慢增加,在15 ns處達到最大(~6000),而后會緩慢降低至4000左右.而抑制劑VVIA和LPFFD與Aβ42之間的接觸數在模擬的初始階段增加比較平緩,且兩種抑制劑和Aβ42之間的接觸數量相當.40 ns后,LPFFD和Aβ42的接觸數大于VVIA和Aβ42之間的接觸數.在模擬的后期, LPFFD和Aβ42之間的接觸數繼續增加到3600以上.圖5為三種多肽抑制劑在80 ns時結合Aβ42的典型構象.從該圖可以看出,三種抑制劑除了和Aβ42發生直接相互作用外,三種抑制劑自身團聚發生相互作用.因此,如果能夠大大降低多肽抑制劑自身之間的團聚可以有利于多肽抑制劑和Aβ42之間的相互作用,從而大大提高其抑制能力.
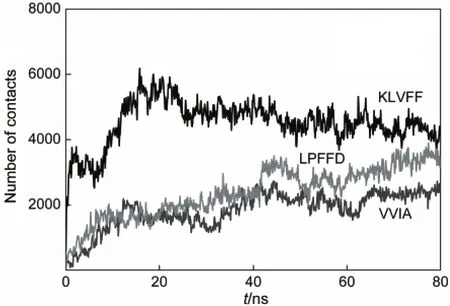
圖4 Aβ42和抑制劑之間的接觸數隨模擬時間的變化Fig.4 Number of contacts betweenAβ42 and peptide inhibitors as a function of simulation time
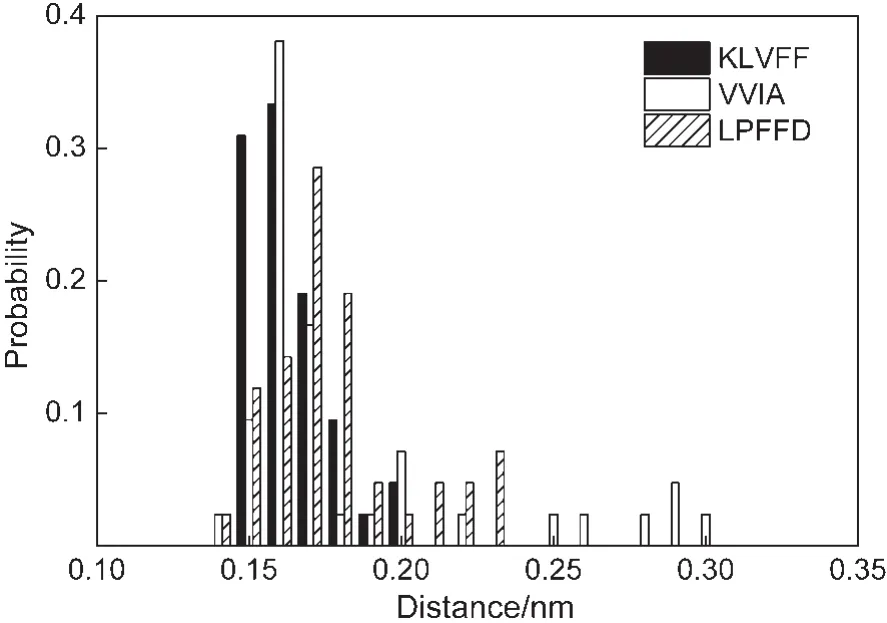
圖6 Aβ42和抑制劑之間的平均接觸距離的概率分布Fig.6 Distribution of the average contact distances betweenAβ42 and peptide inhibitors
圖6為Aβ42和抑制劑之間的平均接觸距離的概率分布.從該圖可以看出在抑制劑KLVFF存在下,Aβ42中氨基酸殘基與抑制劑間的距離分布范圍較小,主要位于0.13-0.18 nm,且分布的峰值位于0.13-0.17 nm之間.說明KLVFF與Aβ42所有殘基之間的作用情況較穩定,作用區域分布無特異性,抑制劑分子分布在Aβ42的整個表面,如圖5A所示. Aβ42與抑制劑LPFFD的接觸距離的概率分布的范圍較寬,峰值位于0.14-0.18 nm.與抑制劑KLVFF相比較,峰值向遠距離方向移動,且概率值分布較平均,說明抑制劑LPFFD與Aβ42之間結合的緊密程度比抑制劑KLVFF差(圖5C).主要原因是脯氨酸的引入破壞了該肽段的線性結構,即僅有三個殘基FFD能夠與Aβ42發生相互作用,從而大大降低了LPFFD和Aβ42之間的結合作用.從而使抑制劑LPFFD和Aβ42之間的接觸距離比抑制劑KLVFF長.抑制劑VVIA和Aβ42的氨基酸之間的接觸距離的最高峰雖然也位于0.16 nm,但其整體概率分布范圍很寬,幾乎遍布0.13-0.27 nm,個別殘基與抑制劑之間的距離達到0.27 nm.這說明抑制劑與部分氨基酸之間的作用力較弱,或不易結合,其原因可能有以下兩點:其一,抑制劑VVIA為四肽而另外兩種抑制劑為五肽,序列較短使得抑制劑無法覆蓋Aβ42分子的整個序列,如圖5B所示;其二,抑制劑VVIA全部由疏水性殘基組成,在模擬過程中出現了明顯的抑制劑自聚現象(圖5B),從而影響了抑制劑與Aβ 42之間的結合,進而影響了VVIA的抑制效果.相反,由于抑制劑KLVFF和LPFFD中含有帶電氨基酸,從而大大降低了它們的自聚能力.綜上所述,抑制劑KLVFF與Aβ42之間的作用力要強于另外兩種多肽類抑制劑VVIA和LPFFD.
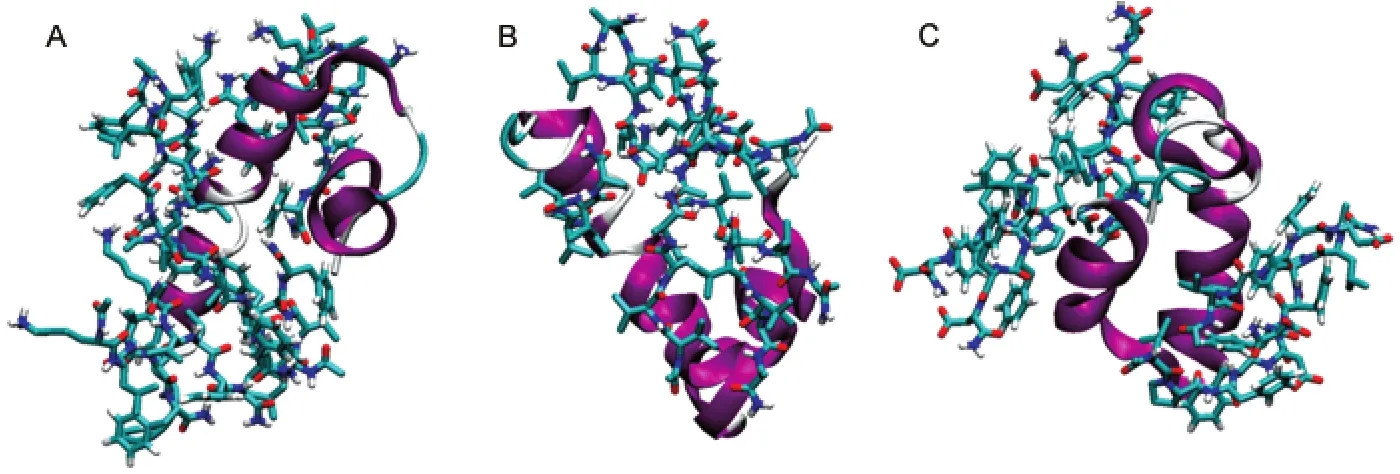
圖5 80 ns時Aβ42和多肽抑制劑之間相互作用的典型構象Fig.5 Representative structures of interaction ofAβ42 and peptide inhibitors at 80 ns(A)KLVFF,(B)VVIA,(C)LPFFD.The three snapshots extracted from the corresponding MD trajectories.For clarity,water molecules are not shown.The main chain ofAβ42 is shown by a NewCartoon model,and the peptide inhibitors are represented by a Licorice model.
3.4 抑制劑和Aβ42之間的相對結合自由能分析
為了進一步解析三種抑制劑抑制Aβ42構象轉換的能力,作者利用MM-PBSA方法計算了抑制劑和Aβ42之間的結合自由能.利用模擬過程平衡階段的最后10 ns(70-80 ns)的模擬軌跡進行分析,并根據分子間作用能(范德華作用能和靜電作用能)和溶劑化作用能(溶劑化非極性作用能和溶劑化靜電作用能)對多肽抑制劑-Aβ42的結合自由能進行分解,結果如表2所示.本文的主要目的是分析比較三種多肽抑制劑抑制Aβ42構象轉換的能力.而計算結合自由能時所用的抑制劑-Aβ42復合物的結構是從分子動力學模擬后期穩定階段(70-80 ns)獲得的.此時,抑制劑和Aβ42的構象基本沒有什么變化.因此,抑制劑和Aβ42之間的分子間作用能和溶劑化作用能對于多肽抑制劑抑制Aβ42構象轉換的能力密切相關,而熵的關系不大.例如,Viet等30計算所得的多肽抑制劑KLVFF和LPFFD與Aβ40之間的熵基本相等,均為138.2 kJ·mol-1·K-1.該現象也被很多研究發現.45因此多肽抑制劑-Aβ42之間熵的貢獻沒有計算.本文中多肽抑制劑和Aβ42之間的自由能為相對結合自由能(忽略熵),而不是絕對結合自由能.另外,已有大量文獻表明,相對結合自由能完全能夠用來表征分子之間的結合作用力強弱.46-49從表2可以看出三種抑制劑和Aβ42之間的非極性相互作用能和靜電相互作用能均為負值,說明在模擬過程中,Aβ42和多肽抑制劑之間的疏水和靜電相互作用均有利于抑制劑和Aβ42結合,從而抑制Aβ42的構象轉換.
從表2可以看出Aβ42與抑制劑KLVFF形成復合物的ΔGbind值最低,為-494.0 kJ·mol-1,說明Aβ42與KLVFF的結合作用最強,結合體最穩定.而與VVIA和LPFFD形成復合物時產生的ΔGbind相似.說明這兩種抑制劑抑制Aβ42的構象轉換能力相差不大.上述結果與側鏈接觸(圖3)以及抑制劑和Aβ42之間的接觸數(圖4)和接觸距離(圖6)分析的結果一致.

表2 多肽抑制劑-Aβ42復合物的結合自由能分解Table 2 Binding free energy components of peptide inhibitors-Aβ42 complex
對比表中各種作用能發現,ΔEvdW,ΔGnps和ΔEelec均為負值,說明多肽抑制劑和Aβ42之間的范德華作用、非極性溶劑化自由能和靜電作用對多肽抑制劑和Aβ42之間的結合均起促進作用.Aβ42與抑制劑KLVFF形成復合物時,ΔGPBelec在ΔGbind中占70%,因此靜電作用在抑制劑KLVFF結合Aβ42的貢獻大.而抑制劑LPFFD與Aβ42形成復合物時,ΔGnonpolar在ΔGbind中占主要地位(>78%),即疏水作用對抑制劑LPFFD與Aβ42之間的結合能的貢獻大.對比抑制劑KLVFF和LPFFD與Aβ42之間的靜電相互作用能可以發現,KLVFF和Aβ42之間的靜電相互作用遠大于LPFFD.主要是因為抑制劑KLVFF中的賴氨酸(K)帶正電荷,可以與Aβ42上的帶負電荷的氨基酸殘基(3個天冬氨酸和3個谷氨酸殘基)產生較強的靜電相互作用.此外,帶負電荷的氨基酸殘基較多且分布較廣,從而有利于和抑制劑KLVFF中的賴氨酸發生作用.因此Aβ42和KLVFF之間的靜電作用較大.與此相反,抑制劑LPFFD帶負電荷,雖然可以與Aβ42上的帶正電荷的氨基酸殘基(1個精氨酸和2個賴氨酸殘基)產生較強的靜電相互作用.但由于Aβ42中的帶正電荷的氨基酸分布集中,且Aβ42整體也帶負電荷,使得LPFFD和Aβ42之間的靜電作用能小于KLVFF.
綜上所述,多肽抑制劑與Aβ42之間的靜電和疏水相互作用均有利于它們之間的結合.抑制劑KLVFF與Aβ42結合過程中分子間作用以靜電作用為主,而抑制劑VVIA和LPFFD與Aβ42之間的作用以疏水相互作用為主.
3.5 抑制劑和Aβ42之間的氫鍵分析
早期研究表明,Aβ42構象轉換的推動力主要有疏水和靜電(包括氫鍵)相互作用.29此外氫鍵網絡還是維持寡聚體和纖維規則結構的重要作用形式.50圖7顯示了Aβ42和三種抑制劑之間的氫鍵數量隨模擬時間的變化.比較三種不同的抑制劑發現,Aβ42與抑制劑KLVFF相互作用時生成的氫鍵個數明顯多于與另兩種抑制劑之間產生的氫鍵個數.在模擬的初始階段抑制劑KLVFF和Aβ42之間的氫鍵個數迅速上升,在25-65 ns期間氫鍵達到13個左右,65 ns后氫鍵數量略有下降,最終氫鍵個數維持在12個左右.抑制劑VVIA和LPFFD與Aβ42相互作用形成的氫鍵個數在整個模擬過程中緩慢增長,到70 ns后VVIA與Aβ42之間的氫鍵個數趨于穩定(約7個),而LPFFD和Aβ42之間的氫鍵個數在5個以下.
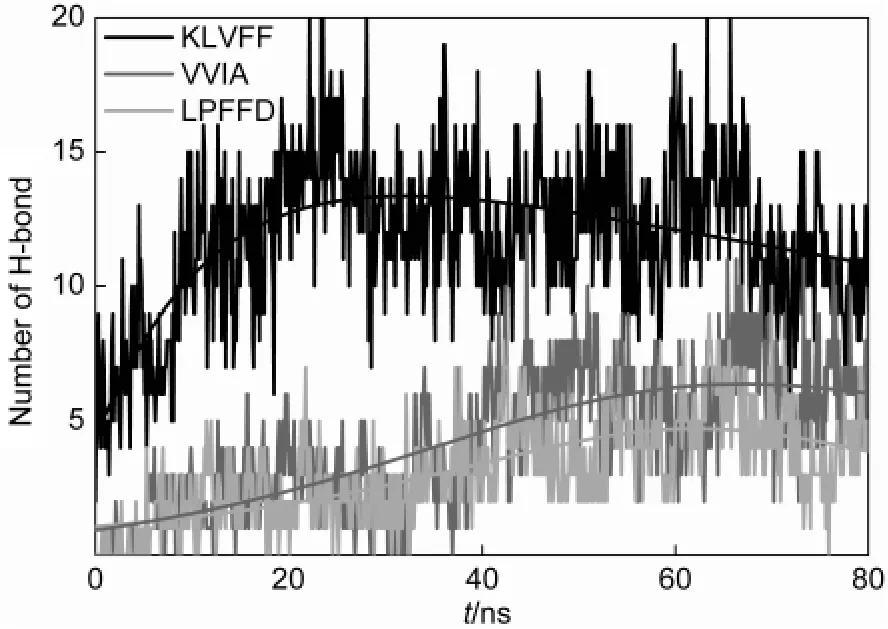
圖7 Aβ42和抑制劑之間的氫鍵數量隨模擬時間的變化Fig.7 Number of H-bonds between peptide inhibitors and Aβ42 as a function of simulation timeData are fitted by a logarithm function.
KLVFF是Aβ42的疏水核心,大量的實驗和分子模擬結果顯示它與Aβ42具有非常強的結合能力.30,51KLVFF含有四個疏水性殘基和一個帶電殘基.該抑制劑除了通過主鏈的羰基和亞氨基與Aβ42形成氫鍵外,帶電殘基K含有較多的氫鍵供體和受體,它與Aβ42之間也能形成大量的氫鍵.相反,抑制劑VVIA的所有殘基均為疏水性氨基酸,它們只能通過主鏈的羰基和亞氨基與Aβ42之間形成氫鍵,所以它與Aβ42之間的氫鍵數量要小于KLVFF.抑制劑LPFFD是KLVFF的衍生物,其氨基酸組成和KLVFF類似,均含有四個疏水性殘基和一個帶電殘基.其與Aβ42之間的氫鍵數量要遠小于KLVFF的主要原因是脯氨酸(P)的引入,破壞了該肽段的線性結構(圖1D).從而使其與Aβ42之間的氫鍵數遠小于KLVFF.在抑制劑LPFFD中的脯氨酸,可以抑制其它Aβ分子繼續聚集,但同時也降低了其與Aβ的結合能力.綜上所述,多肽抑制劑和Aβ42形成氫鍵數量的能力與其抑制Aβ42構象轉換的能力正相關.多肽抑制劑中含有帶電殘基有利于其與Aβ42形成氫鍵,從而更有利于抑制其構象轉換和聚集.
4 結論
利用全原子分子動力學模擬方法考察了三種多肽抑制劑KLVFF、VVIA和LPFFD對Aβ42構象變化的抑制作用,并分析了三種抑制劑與Aβ42結合過程的差異,得到如下結論:三種多肽抑制劑均能夠有效抑制Aβ42的構象變化,穩定其初始的α-螺旋結構,尤其是N端α-螺旋構象,從而阻止了β-折疊構象的生成.多肽抑制劑還使Aβ42分子內遠距離的接觸減少,有效抑制了Aβ42的疏水塌縮,從而降低了Aβ42分子內的疏水相互作用,并起到穩定其初始構象的作用.抑制劑與Aβ42之間的疏水和靜電作用(包括氫鍵)均有利于它們抑制Aβ42的構象轉換.由于抑制劑KLVFF和Aβ42之間的疏水和靜電作用均強于其它兩種多肽抑制劑(VVIA和LPFFD),因此抑制劑KLVFF抑制Aβ42構象轉換的效果最好.另外,抑制劑中含有的帶電氨基酸殘基可以增強其和Aβ42之間的靜電相互作用(包括氫鍵),并降低了抑制劑之間的聚集,從而對Aβ42構象轉換的抑制效果較好.在多肽抑制劑中引入脯氨酸會破壞多肽的線性結構,從而大大降低多肽抑制劑與Aβ42之間的結合作用力.
(1) Yan,H.;Jiang,F.C.Acta Phys.-Chim.Sin.2006,22,359. [鄢 浩,姜鳳超.物理化學學報,2006,22,359.]doi:10.3866/ PKU.WHXB20060321
(2) Jakob-Roetne,R.;Jacobsen,H.Angew.Chem.Int.Edit.2009, 48,3030.doi:10.1002/anie.200802808
(3) Hardy,J.A.;Higgins,G.A.Science 1992,256,184.doi: 10.1126/science.1566067
(4)Luo,Z.W.;Wang,D.D.;Lai,L.H.;Xu,X.J.;Li,C.X.Acta Phys.-Chim.Sin.1995,11,419.[駱兆文,王丹丹,來魯華,徐筱杰,李崇熙.物理化學學報,1995,11,419.]doi:10.3866/ PKU.WHXB19950507
(5)Karran,E.;Mercken,M.;De Strooper,B.Nat.Rev.Drug Discov.2011,10,698.doi:10.1038/nrd3505
(6) Hardy,J.;Selkoe,D.J.Science 2002,297,353.doi:10.1126/ science.1072994
(7) Esler,W.P.;Wolfe,M.S.Science 2001,293,1449.doi:10.1126/ science.1064638
(8) DaSilva,K.A.;Shaw,J.E.;McLaurin,J.Exp.Neurol.2010, 223,311.doi:10.1016/j.expneurol.2009.08.032
(9) Liu,F.F.;Ji,L.;Dong,X.Y.;Sun,Y.J.Phys.Chem.B 2009, 113,11320.doi:10.1021/jp905580j
(10) Xu,Y.;Shen,J.;Luo,X.;Zhu,W.;Chen,K.;Ma,J.;Jiang,H. Proc.Natl.Acad.Sci.U.S.A.2005,102,5403.doi:10.1073/ pnas.0501218102
(11) Rochet,J.C.;Lansbury,P.T.,Jr.Curr.Opin.Struct.Biol.2000, 10,60.doi:10.1016/S0959-440X(99)00049-4
(12) Mason,J.M.;Kokkoni,N.;Stott,K.;Doig,A.J.Curr.Opin. Struct.Biol.2003,13,526.doi:10.1016/S0959-440X(03) 00100-3
(13) Bartolini,M.;Andrisano,V.ChemBioChem 2010,11,1018.doi: 10.1002/cbic.200900666
(14)Mason,J.M.Future Med.Chem.2010,2,1813.doi:10.4155/ fmc.10.259
(15) Salomone,S.;Caraci,F.;Leggio,G.M.;Fedotova,J.;Drago,F. Br.J.Clin.Pharmacol.2012,73,504.doi:10.1111/ j.1365-2125.2011.04134.x
(16) Sciarretta,K.L.;Gordon,D.J.;Meredith,S.C.Methods Enzymol.2006,413,273.doi:10.1016/S0076-6879(06)13015-3
(17) Funke,S.A.;Willbold,D.Curr.Pharm.Des.2012,18,755.doi: 10.2174/138161212799277752
(18) Li,H.;Zemel,R.;Lopes,D.H.;Monien,B.H.;Bitan,G. ChemMedChem 2012,7,515.doi:10.1002/cmdc.201100584
(19) Yang,C.;Zhu,X.;Li,J.;Shi,R.J.Mol.Model.2010,16,813. doi:10.1007/s00894-009-0594-y
(20) Veloso,A.J.;Kerman,K.Bioelectrochemistry 2012,84,49.doi: 10.1016/j.bioelechem.2011.08.007
(21) Jagota,S.;Rajadas,J.Int.J.Pept.Res.Ther.2012,18,53.doi: 10.1007/s10989-011-9278-4
(22) Tjernberg,L.O.;Naslund,J.;Lindqvist,F.;Johansson,J.; Karlstrom,A.R.;Thyberg,J.;Terenius,L.;Nordstedt,C. J.Biol.Chem.1996,271,8545.doi:10.1074/jbc.271.15.8545
(23) Soto,C.;Kindy,M.S.;Baumann,M.;Frangione,B.Biochem. Biophys.Res.Commun.1996,226,672.doi:10.1006/ bbrc.1996.1413
(24) Soto,C.;Sigurdsson,E.M.;Morelli,L.;Kumar,R.A.;Castano, E.M.;Frangione,B.Nat.Med.1998,4,822.doi:10.1038/ nm0798-822
(25) Adessi,C.;Frossard,M.J.;Boissard,C.;Fraga,S.;Bieler,S.; Ruckle,T.;Vilbois,F.;Robinson,S.M.;Mutter,M.;Banks,W. A.;Soto,C.J.Biol.Chem.2003,278,13905.doi:10.1074/jbc. M211976200
(26) Hetenyi,C.;Szabo,Z.;Klement,E.;Datki,Z.;Kortvelyesi,T.; Zarandi,M.;Penke,B.Biochem.Biophys.Res.Commun.2002, 292,931.doi:10.1006/bbrc.2002.6745
(27) Yang,C.;Li,J.Y.;Li,Y.;Zhu,X.L.J.Mol.Struct.-Theochem 2009,895,1.doi:10.1016/j.theochem.2008.10.003
(28)Wei,G.;Jewett,A.I.;Shea,J.E.Phys.Chem.Chem.Phys. 2010,12,3622.
(29) Liu,F.F.;Dong,X.Y.;He,L.;Middelberg,A.P.;Sun,Y. J.Phys.Chem.B 2011,115,11879.doi:10.1021/jp202640b
(30)Viet,M.H.;Ngo,S.T.;Lam,N.S.;Li,M.S.J.Phys.Chem.B 2011,115,7433.doi:10.1021/jp1116728
(31) Fradinger,E.A.;Monien,B.H.;Urbanc,B.;Lomakin,A.;Tan, M.;Li,H.;Spring,S.M.;Condron,M.M.;Cruz,L.;Xie,C.W.; Benedek,G.B.;Bitan,G.Proc.Natl.Acad.Sci.U.S.A.2008, 105,14175.doi:10.1073/pnas.0807163105
(32) Crescenzi,O.;Tomaselli,S.;Guerrini,R.;Salvadori,S.; D?Ursi,A.M.;Temussi,P.A.;Picone,D.Eur.J.Biochem. 2002,269,5642.doi:10.1046/j.1432-1033.2002.03271.x
(33) van Der Spoel,D.;Lindahl,E.;Hess,B.;Groenhof,G.;Mark, A.E.;Berendsen,H.J.J.Comput.Chem.2005,26,1701.doi: 10.1002/jcc.20291
(34) van Gunsteren,W.F.;Billeter,S.R.;Eising,A.A.; Hünenberger,P.H.;Krüger,P.;Mark,A.E.;Scott,W.R.P.; Tironi,I.G.In Biomolecular Simulation:The GROMOS96 Manual and User Guide;Zürich,Switzerland,Groningen, Holland,1996.
(35) Berendsen,H.J.C.;Postma,J.P.M.;van Gunsteren,W.F.; Hermans,J.Intermolecular Forces;Pullmann,B.Ed.;Reidel: Dordecht,Holland,1981.
(36) Darden,T.;York,D.;Pedersen,L.J.Chem.Phys.1993,98, 10089.doi:10.1063/1.464397
(37) Verlet,L.Phys.Rev.1967,159,98.doi:10.1103/PhysRev.159.98
(38) Hess,B.;Berendsen,H.J.C.;Fraaije,J.G.E.M.J.Comput. Chem.1997,18,1463.doi:10.1002/(SICI)1096-987X(199709) 18:12<1463::AID-JCC4>3.0.CO;2-H
(39) Beredsen,H.J.C.;Postma,J.P.M.;van Gunsteren,W.F.;Di Nola,A.;Haak,J.R.J.Chem.Phys.1984,81,3684.doi: 10.1063/1.448118
(40) Kabsch,W.;Sander,C.Biopolymers 1983,22,2577.doi: 10.1002/bip.360221211
(41) Hu,J.P.;Sun,T.G.;Chen,W.Z.;Wang,C.X.Acta Chim.Sin. 2006,64,2079.[胡建平,孫庭廣,陳慰祖,王存新.化學學報,2006,64,2079.]
(42) Kollman,P.A.;Massova,I.;Reyes,C.;Kuhn,B.;Huo,S.; Chong,L.;Lee,M.;Lee,T.;Duan,Y.;Wang,W.;Donini,O.; Cieplak,P.;Srinivasan,J.;Case,D.A.;Cheatham,T.E.,III. Accounts Chem.Res.2000,33,889.doi:10.1021/ar000033j
(43) Li,H.;Luo,Y.;Derreumaux,P.;Wei,G.J.Phys.Chem.B 2010, 114,1004.doi:10.1021/jp908889q
(44) Liu,F.F.;Dong,X.Y.;Sun,Y.Acta Phys.-Chim.Sin.2010,26, 1643.[劉夫鋒,董曉燕,孫 彥.物理化學學報,2010,26, 1643.]
(45)Hu,J.P.;Gong,X.Q.;Su,J.G.;Chen,W.Z.;Wang,C.X. Biophys.Chem.2008,132,69.doi:10.1016/j.bpc.2007.09.008
(46)Huang,B.;Liu,F.F.;Dong,X.Y.;Sun,Y.J.Phys.Chem.B 2012,116,424.doi:10.1021/jp205770p
(47)Huang,B.;Liu,F.F.;Dong,X.Y.;Sun,Y.J.Phys.Chem.B 2011,115,4168.doi:10.1021/jp111216g
(48)Hou,T.;Wang,J.;Li,Y.;Wang,W.J.Comput.Chem.2011,32, 866.doi:10.1002/jcc.21666
(49) Liu,F.F.;Liu,Z.;Bai,S.;Dong,X.Y.;Sun,Y.J.Chem.Phys. 2012,136,145101.doi:10.1063/1.3702195
(50) Santini,S.;Wei,G.;Mousseau,N.;Derreumaux,P.Structure 2004,12,1245.doi:10.1016/j.str.2004.04.018
(51)Kokkoni,N.;Stott,K.;Amijee,H.;Mason,J.M.;Doig,A.J. Biochemistry 2006,45,9906.doi:10.1021/bi060837s
May 14,2012;Revised:July 16,2012;Published on Web:July 16,2012.
Molecular Dynamics Simulation and Binding Free Energy Calculation of the Conformational Transition of Amyloid Peptide 42 Inhibited by Peptide Inhibitors
DONG Xiao-Yan DU Wen-Jie LIU Fu-Feng*
(Department of Biochemical Engineering,School of Chemical Engineering and Technology,Tianjin University,Tianjin 300072,P.R. China; Key Laboratory of Systems Bioengineering,Ministry of Education,Tianjin University,Tianjin 300072,P.R.China)
The molecular mechanisms of the conformational transition of amyloid β-peptide(Aβ)42 inhibited by the peptide inhibitors KLVFF,VVIA,and LPFFD were studied by using molecular dynamics simulations and binding free energy calculations.These studies confirmed that the conformational transition of Aβ42 from its initial α-helix to β-sheet structure is prevented by these three peptide inhibitors. The calculations also demonstrated that the intra-peptide hydrophobic interactions of Aβ42 are weakened, and its quantity of long range contacts decreased by these inhibitors.Consequently,the hydrophobic collapse of Aβ42 is alleviated and its initial structure is maintained well.Both hydrophobic and electrostatic interactions,including hydrogen bonding,were found to favor the binding of these peptide inhibitors to Aβ42.Moreover,the charged residues of the inhibitors were shown to enhance the electrostatic interactions including hydrogen bonding,decreasing the capacity of the peptide for self-assembly,and increasing the inhibition effect.It was also determined that interactions between the inhibitors and Aβ42 are reduced when proline residue is introduced into the peptide inhibitor,since its linear structure is disrupted. In general,this work has allowed a better understanding of the molecular mechanisms of the effects of the peptide inhibitors KLVFF,VVIA,and LPFFD on the conformational transition of Aβ42 and will assist in the systematic design of high efficiency peptide inhibitors of Aβ aggregation.
Molecular dynamics simulation;Alzheimer?s disease;Amyloid peptide;Peptide inhibitor;Conformational transition
10.3866/PKU.WHXB201207162
?Corresponding author.Email:fufengliu@tju.edu.cn;Tel:+86-22-27406590.
The project was supported by the National Natural Science Foundation of China(20906068,21076149),National Key Basic Research Program of China(973)(2009CB724705),and Natural Science Foundation of Tianjin from Tianjin Municipal Science and Technology Commission,China (10JCYBJC04500).
國家自然科學基金(20906068,21076149),國家重點基礎研究發展規劃項目(973)(2009CB724705)和天津市科委自然科學基金(10JCYBJC04500)資助
O641
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50