藏藥婆婆納藥材薄層鑒別方法學研究
2012-12-23 05:27:00王云紅張小梅嚴倩茹楊榮平
天然產物研究與開發 2012年1期
王云紅,張小梅,林 芳,胡 榮,嚴倩茹,楊榮平*
1重慶市中藥研究院重慶市中藥資源學重點實驗室,重慶400065;2 成都中醫藥大學,成都611137
婆婆納是常用的藏藥之一,藏藥名冬那端赤,為玄參科婆婆納屬植物長果婆婆納Veronica ciliata Fiseh 的干燥全草。具有清熱,愈瘡,生肌,止血的功效,主要用于治療瘡癤,創傷,炎癥。婆婆納被收載在1995 年版《藏藥部頒》中,但由于婆婆納的標準僅有性狀描述,沒有鑒別,不能很好的控制婆婆納的質量。為能有效控制婆婆納藥材的質量,本研究首次對婆婆納的薄層鑒別進行了系統研究,建立了婆婆納對照藥材薄層鑒別的方法,為婆婆納藥材質量控制提供科學依據。
近年來,婆婆納的研究較少,其有效成分尚不清楚,特征成分不明確,選擇單一成分的對照品進行薄層鑒別沒有依據。因而,本研究通過建立對照藥材的方法對婆婆納藥材進行薄層定性研究。
1 儀器與試藥
1.1 儀器
BP121S 電子天平(萬分之一,北京賽多利斯科學儀器有限公司);BUG25-12 超聲波清洗機(220W,50KHZ,必能信超聲(上海)有限公司);數顯恒溫水浴鍋(上海梅香儀器有限公司);高效硅膠G 薄層板,規格:100 ×100 mm、100 ×200 mm,厚度:0.20~0.25 mm,批號:20101102,青島海洋化工分廠;薄層色譜展開缸(雙槽,規格:100 ×200 mm、200 ×200 mm,上海信誼儀器廠)。
1.2 試藥及藥材
所用試劑均為分析純(重慶川東(集團)有限公司化學試劑廠);
婆婆納對照藥材為西藏自治區食品藥品檢驗所提供的基地生產對口藥材;
婆婆納藥材樣品為西藏自治區食品藥品檢驗所提供的不同批次的樣品。
2 方法與結果
2.1 婆婆納對照藥材薄層條件的建立
2.1.1 提取溶媒的考察
取婆婆納對照藥材3 g(5 份),精密稱定,置于錐形瓶中,分別加乙醚、乙酸乙酯、三氯甲烷、95%乙醇、甲醇40 mL,超聲處理30 min,濾過,濾液蒸干,殘渣加相應的提取液2 mL 使溶解,作為供試品溶液。照薄層色譜法(《中國藥典》2010 年版一部附錄IV B)試驗,分別吸取供試品溶液5 μL,點于同一硅膠G 薄層板上,以環己烷-丙酮(7 ︰3)為展開劑,展開,取出,晾干,噴以10%硫酸乙醇溶液,在110℃加熱至斑點顯色清晰,并置于日光下檢視。結果表明:以甲醇為溶媒提取的供試品斑點較少,反映藥材的信息量少;95%乙醇為溶媒提取的樣品斑點較乙醚等提取樣品的斑點少,且95%乙醇提取的樣品粘度較大,點樣過程有困難;乙醚、乙酸乙酯、三氯甲烷為溶媒提取的樣品斑點信息相似,但乙酸乙酯相對其他兩種有機試劑毒性較小。且乙酸乙酯提取的樣品展開后斑點較多,斑點清晰,分離度好,且乙酸乙酯毒性較小,擬采用乙酸乙酯作為提取溶媒。
2.1.2 顯色劑的考察
取2.1.1 項下的5 個供試品溶液,照薄層色譜法試驗,分別吸取供試品溶液5 μL,點于同一硅膠G 薄層板上,以環己烷-丙酮(7 ︰3)為展開劑,三張薄層板同時展開,取出,晾干,分別噴以10%硫酸乙醇溶液、5%香草醛硫酸溶液、磷鉬酸溶液,在110 ℃加熱至斑點顯色清晰,于日光下檢視,薄層色譜圖見圖1。結果表明:在日光下檢視,10%硫酸乙醇溶液顯色后斑點較清晰,顏色鮮明;5%香草醛硫酸溶液顯色后斑點顏色雖然鮮明,但是顯示出的斑點較少;磷鉬酸溶液顯色后斑點雖然清晰,但是不能反映出斑點的顏色。因而,采用10%硫酸乙醇溶液為顯色劑。

圖1 婆婆納對照藥材薄層鑒別顯色劑的考察Fig.1 chromogenic reagents test of Veronica cciliata Fiseh
2.1.3 提取方法的考察
取婆婆納對照藥材3 g(2 份),精密稱定,置于錐形瓶中,加乙酸乙酯40 mL,分別超聲處理30 分鐘和加熱回流處理30 分鐘,濾過,濾液蒸干,殘渣各加乙酸乙酯2 mL 使溶解,作為供試品溶液。照薄層色譜法試驗,取不同提取方法提取的供試品溶液5 μL,點于同一硅膠G 薄層板上,以環己烷-乙酸乙酯(7 ︰3)為展開劑,展開,取出,晾干,噴以10%硫酸乙醇溶液,置110 ℃烘箱中顯色至斑點清晰,于日光下檢視。結果超聲和加熱回流兩種處理方法的結果差別不大。因此,選用操作簡便的超聲處理為供試品的提取方法。
2.1.4 提取時間的考察
取婆婆納對照藥材3 g,精密稱定,置于錐形瓶內,加乙酸乙酯40 mL,分別超聲處理10、20、30、40 min,濾過,濾液蒸干,殘渣加乙酸乙酯2 mL 使溶解,作為供試品溶液。照薄層色譜法試驗,取不同提取時間提取的供試品溶液5 μL,點于同一硅膠G 薄層板上,以環己烷-乙酸乙酯(7∶3)為展開劑,展開,取出,晾干,噴以10%硫酸乙醇溶液,置110 ℃烘箱中顯色至斑點清晰,于日光下檢視。結果超聲處理30和40 min 的樣品斑點較超聲處理10 和20 min 的樣品斑點多,且較之更清晰。因而,選用超聲處理30 min 為樣品提取的時間。
2.1.5 展開劑選擇
對照藥材溶液的制備:取本品粉末3 g,精密稱定,置于錐形瓶內,加乙酸乙酯40 mL,分別超聲處理30 min,濾過,濾液蒸干,殘渣加乙酸乙酯2 mL 使溶解,作為對照藥材溶液。照薄層色譜法試驗,吸取對照藥材溶液6 μL,點于硅膠G 板上,分別以(1)環己烷-丙酮(6∶4),(2)環己烷-乙酸乙酯(7∶3),為展開劑,展開,取出,晾干,噴以10%硫酸乙醇溶液,置110 ℃烘箱中加熱至斑點顯色清晰,日光下檢視,結果:展開劑(1)(2)所展開的對照藥材色譜斑點分離度均較好,但展開劑(1)展開后的供試品分離效果較好,斑點清晰,顏色鮮艷,因而確定環己烷-丙酮(6∶4)為展開劑。
2.1.6 點樣量考察
吸取2.1.5 項下制備的對照藥材溶液各4、6、8、10、12 μL 分別點于同一硅膠G 薄層板上,以環己烷-丙酮(6 ∶4)為展開劑,展開,取出,晾干,噴以10%硫酸乙醇溶液,加熱至斑點顯色清晰,結果:點樣量為6 μL 時斑點已清晰。因此,確定對照藥材溶液點樣量為6 μL。
2.1.7 方法耐用性實驗
預平衡和預飽和方式的考察:照薄層色譜法,吸取2.1.5 項下制備的對照藥材溶液6 μL,點于硅膠G 薄層板上,以環己烷-丙酮(6∶4)為展開劑,不進行預平衡和預飽和操作,展開,取出,晾干,噴以10%硫酸乙醇溶液,置110 ℃烘箱中顯色至斑點清晰,于日光下檢視。結果未經預平衡和預飽和操作得到的薄層色譜圖分離度較好,斑點清晰,無明顯的邊緣效應。因此,本薄層鑒別不需預平衡和預飽和。
展開溫度和濕度的考察:照薄層色譜法,吸取上述已制備的對照藥材溶液6 μL,點于硅膠G 薄層板上,以環己烷-丙酮(6∶4)為展開劑,在濕度為77%時分別于20、40 ℃環境下及在溫度為20 ℃時分別于濕度為77%、32%環境下展開,取出,晾干,噴以10%硫酸乙醇溶液,置110 ℃烘箱中顯色至斑點清晰,于日光下檢視,結果,溫度、濕度對分離效果無顯著影響。因此認為溫度和濕度對其沒有影響,在通常條件下操作即可。
展開距離的考察:照薄層色譜法,吸取上述已制備的對照藥材溶液6 μL,點于硅膠G 薄層板上,以環己烷-丙酮(6∶4)為展開劑,展開,展開距離分別為8 cm 和11 cm,取出,晾干,噴以10%硫酸乙醇溶液,置110 ℃烘箱中顯色至斑點清晰,于日光下檢視。結果:展開距離為8 cm 時對照藥材已能較好的分離,但展距為11 cm 時效果更好。因此,確定展距在11 cm 左右。
不同薄層板的考察:照薄層色譜法,吸取上述已制備的對照藥材溶液6 μL,分別點于預制硅膠G 薄層板(青島海洋化工廠分廠)和實驗室自制硅膠G薄層板上,以環己烷-丙酮(6∶4)為展開劑,展開,取出,晾干,噴以10%硫酸乙醇溶液,置110 ℃烘箱中顯色至斑點清晰,于日光下檢視。結果:青島海洋化工廠分廠生產的預制板和本實驗自制的硅膠G 薄層板,對供試品溶液均能起到很好的分離效果,說明該鑒別方法的耐用性好,但相比之下,預制板分離效果更好。
2.1.8 對照藥材薄層色譜圖
分別吸取上述已制備的對照藥材溶液6 μL,點于同一硅膠G 薄層板上,按照擬定的薄層鑒別的方法,展開,晾干,檢視。薄層色譜圖見圖2。結果,婆婆納對照藥材可被分離出7 個清晰的斑點,做為特征斑點。
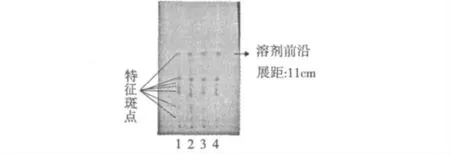
圖2 婆婆納對照藥材薄層色譜圖Fig.2 TLC of Veronica cciliata Fiseh
2.2 婆婆納藥材的薄層鑒別
2.2.1 婆婆納供試品溶液的制備
取本品粉末3 g,加乙酸乙酯40 mL,超聲處理30 分鐘,濾過,濾液蒸干,殘渣加乙酸乙酯2 mL 使溶解,作為供試品溶液。
2.2.2 婆婆納對照藥材的制備
取婆婆納對照藥材3 g,加乙酸乙酯40 mL,超聲處理30 min,濾過,濾液蒸干,殘渣加乙酸乙酯2 mL 使溶解,作為對照藥材溶液。
2.2.3 薄層色譜條件及結果
色譜條件:吸附劑:硅膠G 薄層板;展開劑:環己烷-丙酮(6∶4);顯色劑:10%硫酸乙醇溶液;顯色條件:110 ℃烘箱中加熱至斑點顯色清晰,日光下檢視;點樣量:6 μL。
樣品薄層色譜圖:按照2.2.3(1)項下的色譜條件,將10 批樣品的供試品溶液及對照藥材溶液點于同一薄層板上,展開,晾干,顯色,薄層色譜圖見圖3。結果:10 批樣品在與對照藥材色譜特征斑點相應的位置上均顯相同顏色的斑點。結果表明:建立的方法分離度高,重現性好,簡便,可用于婆婆納的質量控制。
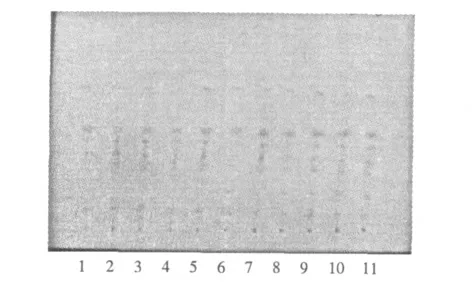
圖3 10 批婆婆納藥材薄層色譜圖Fig.3 TLC of Veronica cciliata Fiseh
3 討論
目前,對婆婆納的研究較少,其有效部位尚不明確,在以后的工作中將進一步研究婆婆納的有效部位,為其質量控制提供依據。本研究以對照藥材為對照,對照藥材主要優點是斑點多,反應的信息多。
藏藥現行標準為《藏藥部頒》1995 年版,很多藥材和制劑,沒有“鑒別”項,或者“鑒別”項下沒有薄層鑒別,僅有理化鑒別或顯微鑒別,不能更好的控制的質量。因而,呼吁有關部門應重視民族藥質量標準的提高,使民族藥這一非物質文化能得以長久流傳。
1 Chinese Pharmacopoeia.2010.
2 People's Republic of China Ministry of health pharmaceutical standards.1995.
