掌葉半夏的HPLC指紋圖譜研究
2013-03-02 03:19:30姜歡欒陽張慧
中國民族民間醫藥 2013年3期
姜 歡 欒 陽 張 慧
遼寧中醫藥大學藥學院,遼寧 大連 116600
掌葉半夏的HPLC指紋圖譜研究
姜 歡 欒 陽 張 慧
遼寧中醫藥大學藥學院,遼寧 大連 116600
目的:采用高效液相色譜法建立掌葉半夏的HPLC色譜指紋圖譜分析方法,為其品質控制提供可靠依據。方法:采用HPLC-UV分析掌葉半夏的指紋圖譜。色譜柱:Kromasil ODS-C18(4.6mm×200mm,5μm)流動相:水(A)-甲醇(B),線性梯度洗脫。體積流量:1.0ml/min。柱溫:25℃;進樣量:10μL。檢測波長260nm。測定7批掌葉半夏指紋圖譜,應用相似度分析、系統聚類分析,對掌葉半夏進行分類研究。結果:在色譜指紋圖譜中,確定了13個共有峰,根據相似度分析、系統聚類分析的結果,將7批藥材分為2類。結論:該方法方便,重現性好,可用于掌葉半夏的質量控制。
掌葉半夏;指紋圖譜;聚類分析
掌葉半夏為天南星科半夏屬植物掌葉半夏Pinellia pedatisecta Schott的干燥塊莖,有燥濕化痰、祛風止痙、消腫散結之功效。其主要化學成分有生物堿[1]、甙類、二肽類、氨基酸等[2]。臨床上應用于宮頸癌治療[3]。藥材混淆品眾多,質量控制指標尚無明確規定。為此,本實驗采用HPLC對不同產地的7批藥材進行了指紋圖譜的研究,建立掌葉半夏較為科學全面的質量評價體系。
1 實驗材料與儀器
1.1 藥材 7批不同產地藥材由安國杏林藥材公司和河北光明藥材有限公司提供,由遼寧中醫藥大學中藥鑒定教研室鑒定為掌葉半夏的塊莖。見表1。
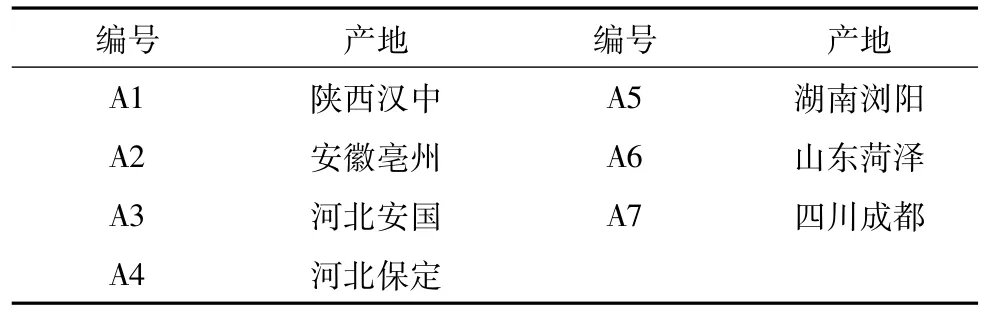
表1 樣品編號及來源
1.2 儀器與試藥 Agilent HPLC1100液相色譜儀,AT-330恒溫柱箱、蒸餾水、甲醇、腺苷對照品。
1.3 色譜條件與適用性試驗 色譜柱:Kromasil ODS-C18(4.6mm×200mm,5μm)
流動相:A:水,B:甲醇。線性梯度5min,A-B(90:10);3min,A-B(90:10);20min,A-B(86:14);30min,A-B(10:90);35min,A-B(0:100);55min,A-B(0:100)。
流速:1.0m l/min。檢測波長260nm。
柱溫:25℃;進樣量:10μL。
記錄時間:75min。
理論塔板數不低于2500。
2 實驗方法
2.1 供試品溶液的制備 取掌葉半夏藥材粉3g(過3號篩),精密稱定,置具塞錐形瓶中,精密加入30%甲醇25m l,精密稱定重量,超聲提取30min,放冷,稱重,加30%甲醇補足減失的重量,0.45μm微孔濾膜濾過,即得。2.2 對照品溶液的制備 精密稱取腺苷對照品7.91mg,置200ml量瓶中,加30%甲醇適量使其溶解,并稀釋至刻度,搖勻,即得。
2.3 測定方法 分別取對照品及供試品溶液經微孔濾膜后,分別精密吸取10μL,注入液相色譜儀,記錄色譜圖。
3 實驗結果
3.1 檢測波長的確定 取樣品溶液,分別在215、254、260、300nm處檢測。結果表明,215nm處基線飄移,300nm各色譜峰之間分離度不好,且峰數不足10個,254、 260nm峰數基本相近,但254nm基線不穩定,不能準確積分,為此確定選用260nm為檢測波長。
3.2 參照物的確定 測定了7個不同產地掌葉半夏藥材樣品,藥材的色譜峰均在60min之內出現,由于對照品腺苷為已知成分,且其為活性成分,其色譜峰在34min左右,不同產地7個樣品均存在,因此選擇此峰為參照物峰。
3.3 共有指紋峰的標定 同時測定了未加掌葉半夏平行制備的陰性樣品溶液,對陰性液與供試品色譜圖進行了比較,排除了陰性干擾峰,以34min出現峰(腺苷)為參照物,選擇7個產地藥材中保留時間與參照物相對比的相對保留時間一致的峰為共有指紋峰,最終確定共有指紋峰13個,經計算,7個產地的各共有峰相對保留時間RSD<3%,每個產地色譜圖中的共有峰的峰面積之和大于90%。見表2。

表2 掌葉半夏藥材指紋圖譜共有峰的峰號
3.4 方法學考察
3.4.1 精密度試驗 由三個不同實驗人員,精密吸取同一供試品溶液10μL,在色譜條件下,重復進樣6次,計算共有指紋峰與參照物的相對保留時間及相對峰面積,結果RSD均小于3.0%,表明精密度良好。
3.4.2 穩定性考察 精密吸取供試品溶液10μL,分別在0、2、4、6、8、10h不同時間點進行測定,以上述各共有指紋峰為檢測指標,計算共有峰的相對保留時間及相對峰面積,結果表明,RSD均小于3.0%,供試品溶液在室溫10h內穩定性良好。
3.4.3 重復性考察 精密稱取山東菏澤掌葉半夏藥材6份,按供試品溶液制備樣品,分別量取10μL注入液相色譜儀,以上述各共有指紋峰為檢測指標,計算共有峰的相對保留時間及峰面積,結果顯示,RSD均小于3.0%,重復性良好。
3.4.4 不同產地掌葉半夏化學成分的指紋圖譜測定 取7個不同產地掌葉半夏藥材粉末,按述供試品溶液制備方法制備樣品,分別量取10μL注入色譜儀,進行測定,計算相對保留時間和相對峰面積,結果顯示:7個產地的13個共有峰的相對保留時間RSD<3%,7個產地的共有峰的相對峰面積相差較大,證明不同產地掌葉半夏藥材質量有一定差異。見表3、4、圖1。
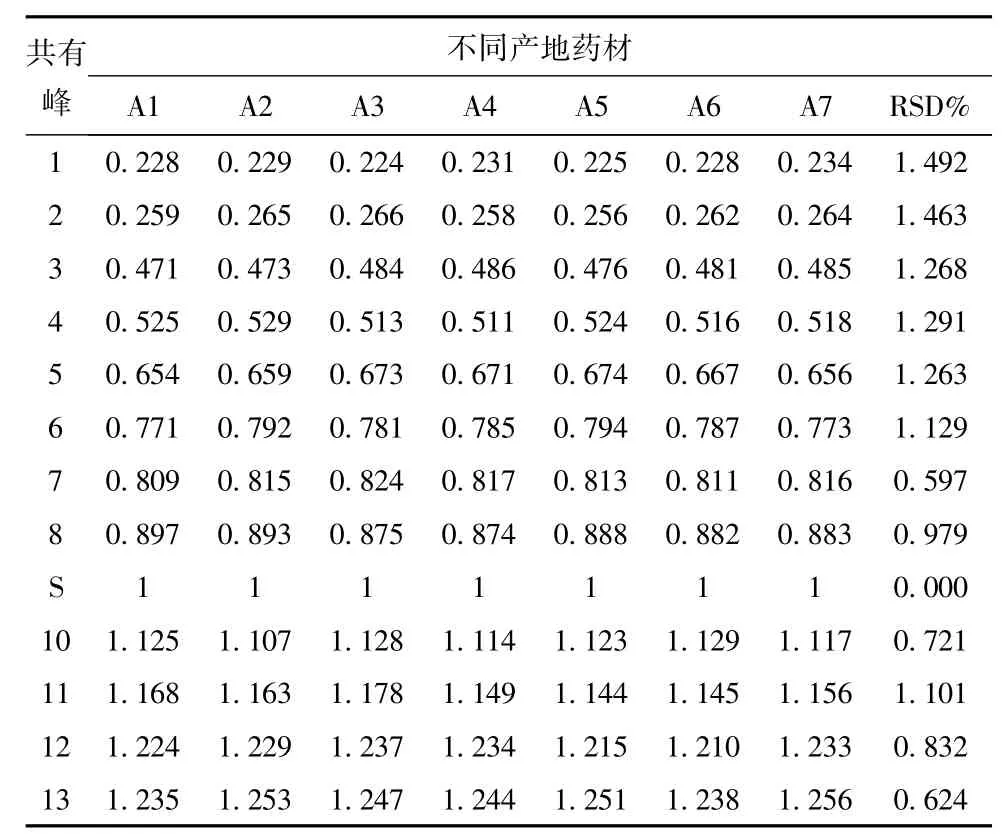
表3 不同產地藥材相對保留時間的指紋圖譜測定結果
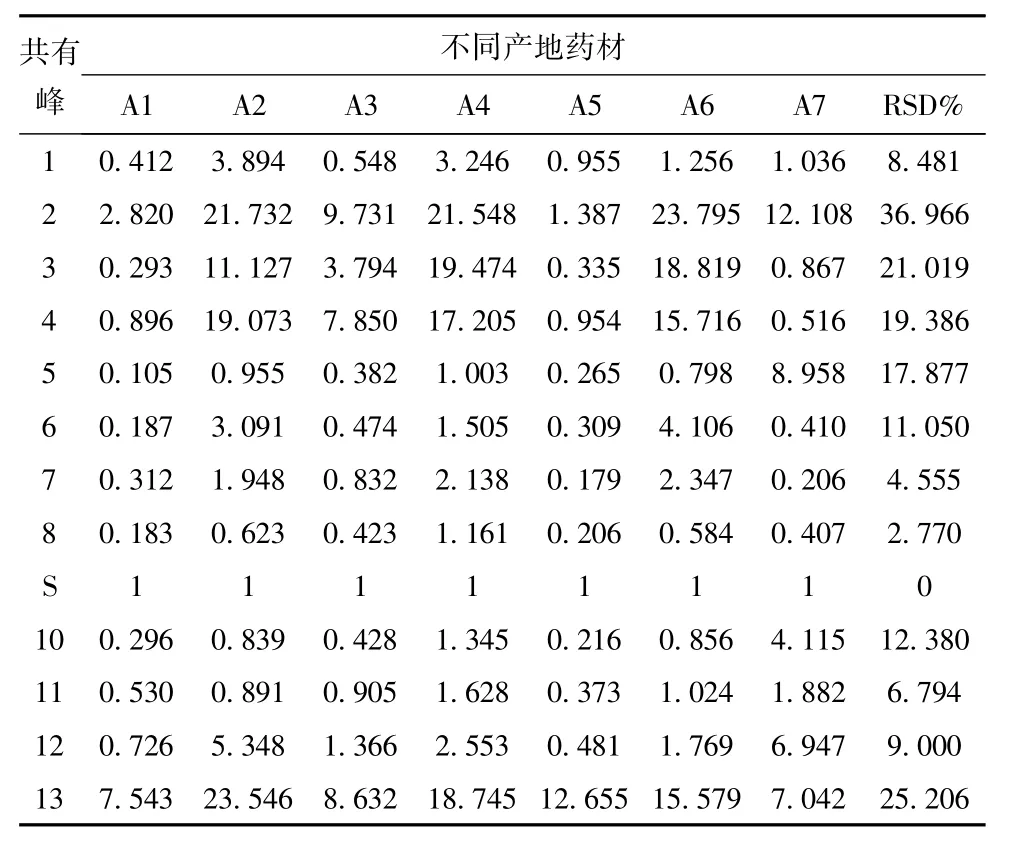
表4 不同產地藥材相對峰面積的指紋圖譜測定結果
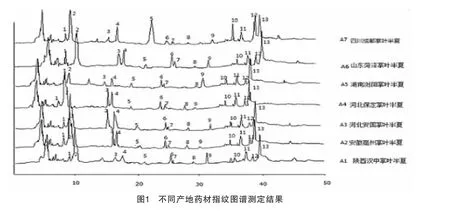
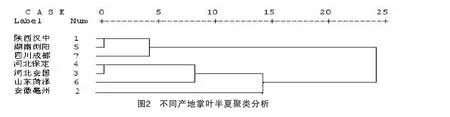
3.4.5 掌葉半夏化學成分指紋圖譜的聚類分析研究 本文采用SPSS軟件對指紋譜圖進行了系統聚類分析,得到聚類樹圖5-11。聚類樹圖顯示:產地A1、A5、A7聚為1類,A4、A3、A6聚為1類,A2聚為1類。見圖2。
4.1 掌葉半夏化學成分指紋圖譜方法的建立 本實驗以腺苷為參照物,采用梯度洗脫的HPLC法,建立了掌葉半夏藥材的指紋圖譜的測定方法,確定了13個共有峰,經精密度、穩定性、重復性等方法學考察,RSD%均小于3.0%,證明此方法可行。
4.2 掌葉半夏化學成分指紋圖譜的聚類分析 7個產地掌葉半夏指紋圖譜的聚類樹圖顯示,產地A1、A5、A7聚為1類,產地A4、A3、A6聚為1類,產地A2與其它6個產地距離較遠,聚為1類,聚類分析結果與藥材的形態學鑒別基本一致,產地相鄰的藥材基本聚為一類。
4.3 小結 本實驗以腺苷為參照物,采用梯度洗脫的HPLC法,建立了掌葉半夏化學成分指紋圖譜。根據7個不同產地掌葉半夏指紋圖譜的相對保留時間,確定了13個共有指紋峰和共有峰位置,并建立標準指紋圖譜的共有模式。采用系統聚類分析方法,建立了掌葉半夏質量評價體系,對7個產地藥材進行了定量分類,聚類分析結果與藥材的形態學鑒別基本一致。
實驗證明,該方法穩定易行,為評價和控制掌葉半夏藥材內在質量提供了科學依據。
[1]秦文娟,王瑞,溫月笙,等.掌葉半夏化學成分的研究(Ⅴ)[J].中草藥,1995,26(1):3.
[2]王瑞,溫月笙,楊嵐,等.掌葉半夏化學成分的研究[J].中國中藥雜志,1997,22(7):421-423.
[3]上海第一醫學院婦產科醫院,基礎部化學教研組.掌葉半夏治療子宮頸癌的研究[J].上海醫學,1978,1(1):13-16.
R284.1
A
1007-8517(2013)03-0034-02
2012.12.29)
