呋布西林鈉合成工藝研究
2013-09-13 13:06:54郭劍虹李祖文周改平
太原理工大學(xué)學(xué)報(bào) 2013年3期
郭劍虹,李祖文,周改平
(山西博康藥業(yè)有限公司,太原030021)
呋布西林鈉,又稱呋脲芐青霉素鈉,其化學(xué)名稱為:(2S,5R,6R)-3,3-二甲基-6-[D-α-(3-(2-呋喃甲酰)脲)苯乙酰氨基]7-氧代-4-硫雜-1-氮雜雙環(huán)[3.2.0]庚烷-2-甲酸鈉鹽。該藥于1969年由美國Brisol &Mayer公司首次研發(fā)成功,國內(nèi)上海醫(yī)藥工業(yè)研究院、上海第四制藥廠及太原制藥廠于1971年進(jìn)行本品的試制,1976年起國內(nèi)首先應(yīng)用于臨床[1]。本品是半合成廣譜抗生素,對(duì)多種革蘭氏陽性菌及革蘭氏陰性菌均有良好抗菌活性,尤其對(duì)綠膿桿菌的作用比羧芐西林強(qiáng)16倍,是最具有應(yīng)用價(jià)值的抗綠膿桿菌的酰脲類抗生素,臨床上主要用于創(chuàng)面感染、敗血癥、呼吸道、泌尿道感染等疾病,用途甚廣。
目前文獻(xiàn)報(bào)道的呋脲芐青霉素鈉的合成路線有3條。路線1是氨芐酸法,由α-呋喃甲酰異氰酸酯與無水氨芐西林三乙胺鹽在二氯甲烷中反應(yīng),經(jīng)提取精制,得到呋脲芐青霉素酸固體,再與碳酸氫鈉于水溶液中成鹽,經(jīng)冷凍干燥制得呋脲芐青霉素鈉[2]。路線2是6-氨基青霉烷酸(6-APA)法,由α-呋喃酰脲基苯乙酸鈉(前體酸鈉)與氯甲酸乙酯在丙酮中反應(yīng)生成混合酸酐,再與6-APA鈉鹽溶液縮合得到呋脲芐青霉素鈉粗品溶液,經(jīng)提取精制后,經(jīng)過冷凍干燥得到呋脲芐青霉素鈉[3]。路線3是將α-呋喃甲酰異氰酸酯與無水氨芐西林在四氫呋喃中反應(yīng),然后加入甲醇鈉-無水乙醇溶液,經(jīng)減壓脫溶劑制得呋脲芐青霉素鈉[4]。上述3條路線各有利弊。路線1的合成步驟復(fù)雜,過程中產(chǎn)生大量的廢水,而且α-呋喃甲酰異氰酸酯的穩(wěn)定性不強(qiáng),特別容易吸濕水解,影響反應(yīng)的進(jìn)行與產(chǎn)品的純度。路線2中的縮合反應(yīng)后處理步驟復(fù)雜,需經(jīng)過堿化、酸化、再堿化的幾次提取,溶劑耗用量大,相轉(zhuǎn)移過程中產(chǎn)生大量廢水,而且不能得到呋布西林酸的固體,液相中的呋布西林酸因含有部分有機(jī)溶劑,在凍干過程中易造成真空泵油的乳化而影響凍干效果。路線3采用溶媒結(jié)晶法,產(chǎn)物的純度略高于路線1,但通過樣品比對(duì),溶媒結(jié)晶法的溶解度低于冷凍干燥法。據(jù)資料記載,由于呋布西林鈉溶解度小,尤其在質(zhì)量分?jǐn)?shù)0.9%生理鹽水和質(zhì)量分?jǐn)?shù)5%葡萄糖溶液中較難溶解,給臨床用藥帶來很大的不便,延緩病人的給藥時(shí)間,并導(dǎo)致護(hù)士對(duì)使用該藥產(chǎn)生抵制情緒[5]。鑒于以上因素,我們綜合考慮收率、純度、溶解度等問題,對(duì)原有工藝路線進(jìn)行改進(jìn),以前體酸鈉為起始原料,經(jīng)酸酐化、6-APA成胺鹽、縮合反應(yīng)制得呋布西林酸固體,然后用碳酸氫鈉水溶液成鹽,經(jīng)脫色、除菌過濾、真空冷凍干燥制得呋布西林鈉。
1 合成路線
呋布西林鈉的合成路線見圖1所示。
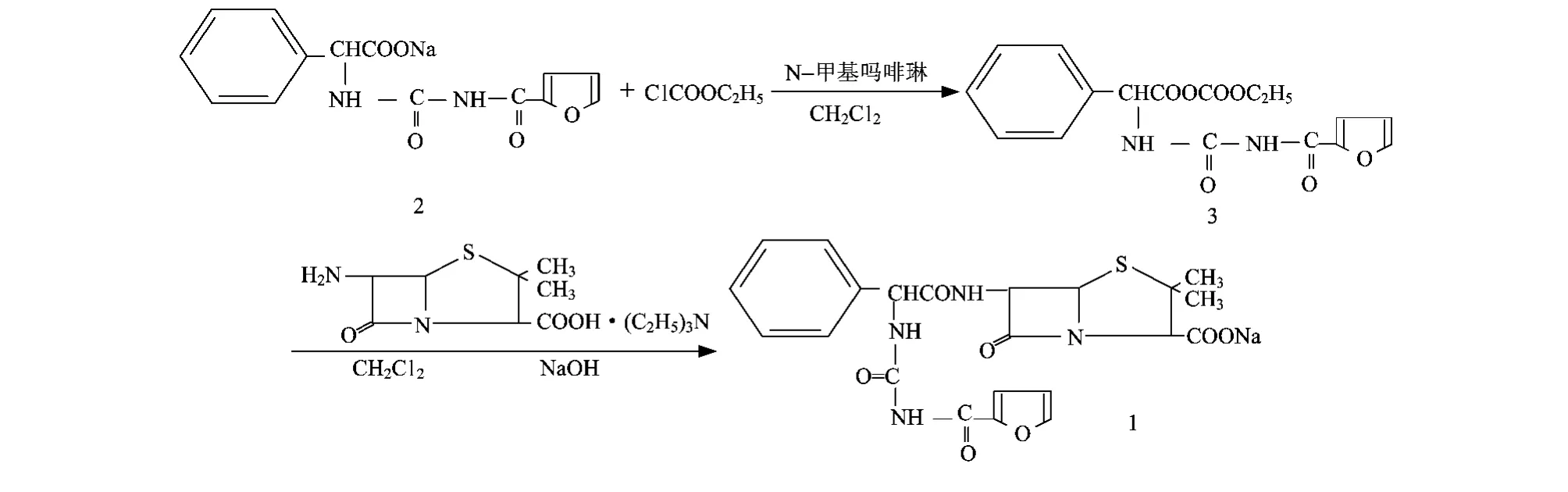
圖1 呋布西林鈉的合成路線
2 實(shí)驗(yàn)部分
2.1 原料與儀器
WZZ-2數(shù)字式自動(dòng)旋光儀(上海浦東物理光學(xué)儀器廠);島津LC-10ATvp型液相色譜儀。6-氨基青霉烷酸(石藥集團(tuán)中諾醫(yī)藥有限公司);α-呋喃甲酰脲基苯乙酸鈉(江蘇正泰醫(yī)藥化工有限公司);氯甲酸乙酯(無錫貝諾精細(xì)化工有限公司);呋布西林鈉對(duì)照品(蘇州二葉制藥有限公司)。鹽酸和碳酸氫鈉為試劑級(jí),其他原料規(guī)格均為工業(yè)級(jí)。
2.2 實(shí)驗(yàn)步驟
1)在干燥的反應(yīng)瓶中加入180mL二氯甲烷和15.0g(0.069 4mol)6-APA,攪拌降溫至10~15℃時(shí)開始滴加34mL三乙胺(大約需要1.5h),直至反應(yīng)液澄清。控制pH值在7.5~8.0之間,得到6-APA胺鹽溶液,降溫至0℃以下備用。
2)在另一干燥的反應(yīng)瓶中加入230mL二氯甲烷,攪拌降溫至-5℃時(shí)加入α-呋喃酰脲基苯乙酸鈉(2)18.7g(0.060 3mol)。繼續(xù)降溫至-10℃,加入N-甲基嗎啡啉0.15mL。5min后加入7.10mL氯甲酸乙酯,于-15~-20℃保溫反應(yīng)1h,然后快速加入6-APA胺鹽溶液。期間溫度會(huì)回升,應(yīng)控制溫度不超過0℃,繼續(xù)降溫至-5℃保溫反應(yīng)30min。
3)將反應(yīng)液升至室溫,用質(zhì)量分?jǐn)?shù)8%的氫氧化鈉水溶液堿化至溶液pH值為5.8~6.5,靜置,溶液分相,回收有機(jī)相。水相中加入150mL醋酸丁酯,經(jīng)質(zhì)量分?jǐn)?shù)9%的鹽酸酸化至pH=2~3,靜置分層,棄去水層。有機(jī)相中細(xì)流加入90mL正己烷(約0.5h),攪拌2.5h.待結(jié)晶完全析出后,過濾,濾餅用純化水洗滌,于50~55℃干燥,得白色結(jié)晶物27.8g。測(cè)得產(chǎn)物收率82.4%,純度94.2%。
4)在反應(yīng)瓶中加入250mL注射用水和25g呋布西林酸,于10~15℃緩慢均勻滴加質(zhì)量分?jǐn)?shù)10%碳酸氫鈉水溶液,調(diào)節(jié)pH值在6.5~6.8之間。待溶液澄清后,測(cè)得pH值在上述范圍內(nèi)。加針劑炭2 g脫色,過濾,進(jìn)行冷凍干燥,得到呋布西林鈉(1)的白色晶體24.9g.測(cè)得二步總收率78.5%(以6-APA計(jì)),純度92.6%(經(jīng) HPLC法測(cè)定)。
3 實(shí)驗(yàn)結(jié)果與討論
3.1 溶劑的影響
文獻(xiàn)[3]中的縮合反應(yīng)在丙酮-水的混合體系中進(jìn)行,由于酸酐忌水,導(dǎo)致部分酸酐水解破壞,影響產(chǎn)物的收率和純度。在對(duì)反應(yīng)液進(jìn)行提純處理的過程中,因體系中的丙酮與水互溶,需要用甲苯萃取丙酮以實(shí)現(xiàn)有機(jī)相與水相的分離。這造成回收溶劑困難,生產(chǎn)成本上升;而且,甲苯屬于毒性化合物,對(duì)皮膚、黏膜有刺激性,短時(shí)間內(nèi)吸入較高濃度可發(fā)生急性中毒導(dǎo)致抽搐或昏迷,長期接觸可發(fā)生神經(jīng)衰弱綜合征、肝腫大、女工月經(jīng)異常等慢性中毒癥狀。本工藝用單一的二氯甲烷做溶劑,使全部反應(yīng)在無水的情況下進(jìn)行,減小了酸酐水解對(duì)產(chǎn)物的影響。另外,二氯甲烷密度大,不溶于水,可以直接與水分相,這大大簡化了處理步驟,也避免了甲苯的毒性對(duì)身體及環(huán)境的傷害。
3.2 6-APA成鹽條件的影響
6-APA的主要結(jié)構(gòu)為β-內(nèi)酰胺環(huán),在堿性條件下極易水解導(dǎo)致酰胺環(huán)的破裂,因此要嚴(yán)格控制體系的水分和pH值。以二氯甲烷取代丙酮-水混合溶劑有效地減小了體系的水分,同時(shí)選用有機(jī)堿三乙胺代替氫氧化鈉水溶液,使得反應(yīng)在無水且酸堿度較溫和的環(huán)境中進(jìn)行。另外溫度越高,β-內(nèi)酰胺環(huán)的穩(wěn)定性越差,為此反應(yīng)液澄清后需要降溫至0℃以下(以不結(jié)冰為宜)放置,且時(shí)間越短越好。
3.3 工藝路線的優(yōu)化
目前呋布西林鈉藥品執(zhí)行的是衛(wèi)生部藥品標(biāo)準(zhǔn)(WS1-C2-0021-89),含量測(cè)定方法為綜合容量法,此法無法分離檢出同分異構(gòu)體雜質(zhì)。根據(jù)文獻(xiàn)報(bào)道,對(duì)市售的呋布西林鈉樣品采用HPLC法測(cè)定含量,發(fā)現(xiàn)有些樣品質(zhì)量分?jǐn)?shù)低于80%,有的甚至只有約60%;其HPLC圖譜中含有一個(gè)較大的雜質(zhì)峰(峰面積約占1/3),通過結(jié)構(gòu)分析,推測(cè)樣品中的主要雜質(zhì)為呋布西林鈉的同分異構(gòu)體,異構(gòu)化位置在6-APA 的 C-6位[6,7]。經(jīng)分析認(rèn)為:一方面,因?yàn)榭s合反應(yīng)在6-APA的6-位氨基上進(jìn)行,若縮合反應(yīng)條件控制不佳,可能引起目標(biāo)產(chǎn)物的異構(gòu)化;另一方面,縮合反應(yīng)后的提純處理是實(shí)現(xiàn)同分異構(gòu)體分離的關(guān)鍵,而萃取結(jié)晶法是實(shí)現(xiàn)有機(jī)同分異構(gòu)體分離的有效方法。于是設(shè)計(jì)了在有機(jī)溶劑中加入萃取劑析出呋布西林酸結(jié)晶的路線。有機(jī)溶劑選用醋酸丁酯,因醋酸乙酯在水中的溶解度大于醋酸丁酯,酸化提取時(shí)分層不徹底,故選擇水中溶解度較小的醋酸丁酯作提取劑。為了選擇萃取劑,考察了石油醚、硅醚、正己烷和環(huán)己烷,結(jié)果萃取效果都不錯(cuò)。從溶劑回收套用的角度考慮,兩種溶劑的沸點(diǎn)相差越多,越易實(shí)現(xiàn)分離,故選擇沸點(diǎn)較低的正己烷做萃取劑。
3.4 正交試驗(yàn)及結(jié)果分析
在合成過程中,影響收率的主要因素通常為物料配比、溫度、時(shí)間等。本文通過正交試驗(yàn)研究了前體酸鈉與6-APA摩爾比、縮合反應(yīng)溫度、縮合反應(yīng)時(shí)間和正己烷用量等四因素對(duì)呋布西林酸收率的影響。試驗(yàn)選用四因素三水平的L9(34)正交試驗(yàn),各因素的三個(gè)水平如表1所示。
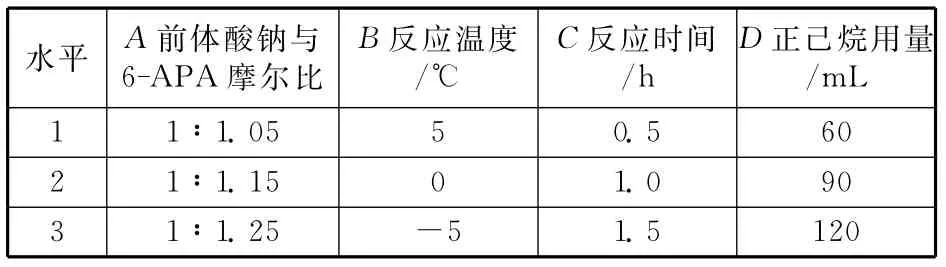
表1 正交試驗(yàn)因素水平表
試驗(yàn)結(jié)果如表2所示。從表2可看出,影響反應(yīng)收率的因素從主到次的順序?yàn)锳、B、D、C;實(shí)驗(yàn)最佳水平組合為A2B3C1D2,即,前體酸鈉與6-APA摩爾比為1∶1.15,反應(yīng)溫度-5℃,反應(yīng)時(shí)間0.5 h,正己烷用量90mL。在此條件下,呋布西林酸的收率達(dá)到82.4%.經(jīng)與碳酸氫鈉成鹽、真空冷凍干燥后,產(chǎn)物的總收率達(dá)到78.5%,純度為92.6%。
3.5 產(chǎn)品表征
用WZZ-2數(shù)字式自動(dòng)旋光儀測(cè)定樣品旋光度,[α]25D為+188°,符合部頒標(biāo)準(zhǔn)(標(biāo)準(zhǔn)為[α]25D=+180°~+200°)。用LC-10ATvp型液相色譜儀作定量分析,色譜條件:C18柱(4.6nm×250mm,5 μm),流動(dòng)相磷酸鹽緩沖液(pH3.5)與乙腈體積比2.5∶1,v=1mL/min,λ=254nm,進(jìn)樣量20μL。測(cè)試結(jié)果顯示,樣品溶液主峰的保留時(shí)間與對(duì)照品溶液主峰的保留時(shí)間一致,樣品純度大于92.6%。
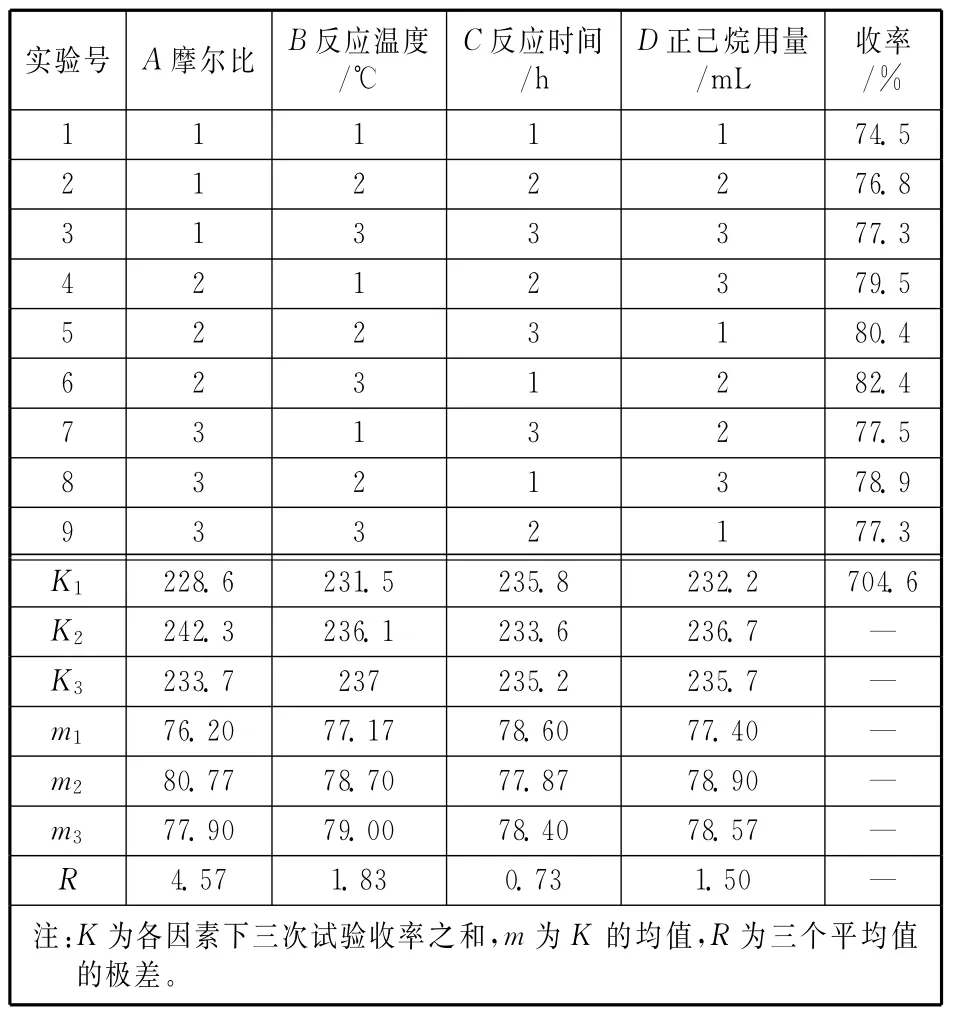
表2 正交試驗(yàn)結(jié)果
對(duì)樣品進(jìn)行溶解性測(cè)試。樣品于18℃時(shí)在注射用水和質(zhì)量分?jǐn)?shù)5%葡萄糖溶液中即溶,在質(zhì)量分?jǐn)?shù)0.9%生理鹽水中3min內(nèi)溶解。
4 結(jié)論
1)本研究的合成使用單相溶劑,從酸酐的制備到與6-APA的縮合反應(yīng)全部為忌水反應(yīng),有效地避免了中間體的水解,提高了產(chǎn)物的收率和純度。
2)以低毒溶劑替代高毒溶劑甲苯,最大限度地改善了工作環(huán)境,同時(shí)還縮短了工藝操作程序,節(jié)約了成本。
3)采用萃取結(jié)晶法提純呋布西林酸固體,很好地去除了雜質(zhì),也減小了有機(jī)溶劑對(duì)凍干機(jī)的損害。
4)通過正交試驗(yàn),確定了最佳反應(yīng)條件,由此獲得的目標(biāo)產(chǎn)物總收率≥78%,純度≥92%;而且溶解性明顯改善,尤其在質(zhì)量分?jǐn)?shù)0.9%生理鹽水和質(zhì)量分?jǐn)?shù)5%葡萄糖溶液中3min內(nèi)溶解,方便了臨床使用。
可見,本工藝收率高、純度高、溶解性強(qiáng)、成本低,很適合工業(yè)化生產(chǎn)。
[1] 張梅芳,顧亞明,張敬德,等.呋芐青霉素的實(shí)驗(yàn)研究[J].抗生素,1985,10(2):85-91.
[2] 李中華,楊峰.呋脲芐西林鈉的合成新工藝[J].太原理工大學(xué)學(xué)報(bào),2004,35(5):575-576.
[3] 馬玉卓,劉鷹翔,周玉平,等.呋布西林鈉的合成工藝改進(jìn)[J].中國藥物化學(xué)雜志,2006,16(1):51-53.
[4] 蔣征昊.制備呋脲芐青霉素鈉的方法:中國,CN1854141A [P].2006-11-01.
[5] 劉力.易溶解的抗菌藥物組合藥物制劑及制備方法:中國,CN1872057A [P].2006-12-06.
[6] 張菁,姜建國,高燕霞.呋芐西林鈉中相關(guān)物質(zhì)的高效液相色譜-電噴霧電離-質(zhì)譜分析[J].中國藥業(yè),2009,18(7):16-17.
[7] 容凱文,于沛,王定遠(yuǎn).注射用呋芐西林鈉中主要雜質(zhì)的結(jié)構(gòu)推測(cè)[J].中國醫(yī)藥工業(yè),2010,41(5):373-374.
