不銹鋼載波氧化膜/聚噻吩復(fù)合膜的制備及耐蝕性能研究
2013-09-14 05:01:28梁成浩黃乃寶
材料工程 2013年1期
關(guān)鍵詞:不銹鋼
梁成浩,陳 婉,黃乃寶
(1大連海事大學(xué) 交通運(yùn)輸裝備與海洋工程學(xué)院,遼寧 大連 116026;2大連理工大學(xué) 化工學(xué)院,遼寧 大連 116023)
電化學(xué)合成的聚噻吩膜具有良好的電化學(xué)穩(wěn)定性,在催化、電化學(xué)傳感器、材料保護(hù)等領(lǐng)域有著廣闊的應(yīng)用前景[1,2]。多數(shù)情況下,聚噻吩膜是在貴金屬上合成,而在不銹鋼、鋁、鈦等氧化性金屬上合成聚噻吩膜既可以節(jié)約成本,又能擴(kuò)展導(dǎo)電高分子膜的應(yīng)用范圍。然而,與在惰性電極上生成的聚噻吩膜相比,在氧化性金屬上形成的膜多疏松且與基體結(jié)合力差。這是由于氧化性金屬產(chǎn)生陽極溶解活性,且陽極溶解電位低于聚噻吩單體的氧化電位。因此,在電氧化聚合過程中,陽極金屬的不穩(wěn)定性阻滯導(dǎo)電高分子膜在電極上的成核過程,導(dǎo)致氧化形成的高分子膜疏松,與基體結(jié)合力欠佳。為解決這一問題,對(duì)金屬基體進(jìn)行表面改性是改善高分子膜與基體結(jié)合力的一個(gè)途徑。Ren等[3]將430鐵素體不銹鋼經(jīng)磷酸鈍化,形成微孔表面后,電沉積聚3-甲基噻吩(PMeT)膜,使不銹鋼基體上成功包覆一層結(jié)合力良好的PMeT膜,并發(fā)現(xiàn)未經(jīng)鈍化的金屬表面無法沉積PMeT膜。對(duì)不銹鋼可采用多種表面改性方法獲得多孔表面,其中,20世紀(jì)90年代開發(fā)的不銹鋼低頻載波氧化法[4,5]是一種有效的方法。它可通過調(diào)節(jié)載波條件控制氧化膜的結(jié)構(gòu)、半導(dǎo)體性能及厚度,克服了傳統(tǒng)方法使用鉻酸鹽的缺點(diǎn),可稱之為綠色工藝。
本工作旨在以304奧氏體不銹鋼為基體,經(jīng)不同條件載波氧化后,電沉積聚噻吩制備了“氧化層/聚噻吩”的復(fù)合膜,并考察了復(fù)合膜的耐蝕性能,為導(dǎo)電高分子復(fù)合膜的研制提供了一個(gè)新的途徑。
1 實(shí)驗(yàn)方法
1.1 304不銹鋼的載波鈍化
304不銹鋼的載波鈍化在70℃的2.5MH2SO4鈍化溶液中進(jìn)行,載波鈍化方法的波形電場示于圖1。Eh與El分別代表方波高電位和方波低電位,T為脈沖寬度。本實(shí)驗(yàn)分別在兩種方波高電位下進(jìn)行載波Eh=1.02V(簡稱 AV1020),Eh=0.8V (簡稱AV800);方波低電位為El=-250mV,脈沖寬度為0.3s,陽極電位與陰極電位時(shí)間比例為2∶1,鈍化時(shí)間為10min。處理后試樣經(jīng)去離子水洗后干燥待用。
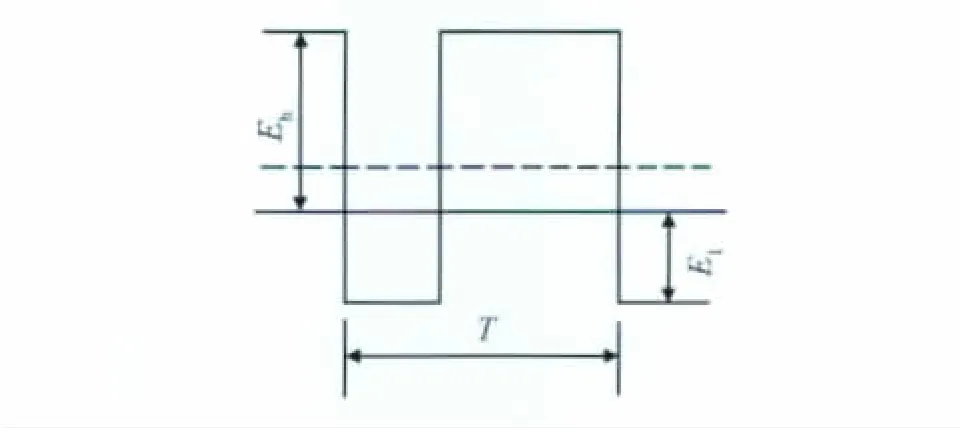
圖1 載波鈍化方波電場示意圖Fig.1 Schematic representation of the square wave electric field
1.2 聚噻吩膜的電化學(xué)沉積
聯(lián)噻吩(99%),東京化成。乙腈(HPLC)經(jīng)4nm分子篩干燥后加入P2O5回流。支持電解質(zhì):六氟磷四丁基胺[tetra(n-butyl ammonium)hexafluorophosphate,TBAPF6],電化學(xué)純,購于Fluka公司。采用恒電流法在304不銹鋼氧化膜上制備聚噻吩膜(簡稱PTH),以玻碳(GC)電極作參照。聚合電流密度控制在0.5mA/cm2。本實(shí)驗(yàn)中PTH膜厚度控制在10μm以下。電解液為5mM聯(lián)噻吩+0.1M的TBAPF6乙腈溶液。聚合反應(yīng)在單池、三電極體系中進(jìn)行。輔助電極為鉑絲,參比電極為 Ag/Ag+(0.1MAgNO3)。電化學(xué)反應(yīng)前,對(duì)加有單體的電解質(zhì)溶液進(jìn)行高純氬氣鼓泡處理,實(shí)驗(yàn)在氬氣保護(hù)下進(jìn)行。
1.3 復(fù)合膜的測試與表征
采用紅外光譜、掃描電鏡與原子力顯微鏡(AFM)對(duì)復(fù)合膜的結(jié)構(gòu)與形貌進(jìn)行表征。使用循環(huán)伏安法對(duì)復(fù)合膜的電化學(xué)性能進(jìn)行測試。將制備好的復(fù)合膜在乙腈溶液浸泡沖洗后,放入單池,以Pt絲為輔助電極,Ag/Ag+(0.1M)為參比電極。在0.1M 的TBAPF6的乙腈溶液中進(jìn)行循環(huán)伏安掃描。循環(huán)伏安電位范圍為-0.5~0.75V,掃描速率為50mV/s。上述實(shí)驗(yàn)前,電解質(zhì)溶液經(jīng)氬氣鼓泡處理15min,并在氬氣保護(hù)下進(jìn)行測試。
按照ASTM—F1044—05標(biāo)準(zhǔn)[6],采用單搭接剪切法測試PTH膜與不銹鋼的界面結(jié)合強(qiáng)度,測試前304不銹鋼板試樣切割成50mm×10mm×2mm的長條狀,搭接重疊區(qū)長度為10mm,實(shí)驗(yàn)前用黏合劑(E-55環(huán)氧樹脂與聚酰胺=1∶1)粘接后,室溫下放置24h。測試儀器為CSS-2205型電子萬能試驗(yàn)機(jī),拉伸速率為0.05mm/min。每組取3個(gè)樣本,取平均值。復(fù)合膜的電導(dǎo)率采用四探針法進(jìn)行測試。
1.4 耐蝕性測試方法
開路電位測試在1M的H2SO4中進(jìn)行,輔助電極為Pt絲,參比電極為SCE。試樣在1MH2SO4中浸泡10min后,以5mV/s的掃描速率從腐蝕電位開始進(jìn)行陽極極化掃描。交流阻抗測試在1M的H2SO4中進(jìn)行,頻率范圍100mHz~100kHz,交流振幅(AC)為10mV。
2 結(jié)果與討論
2.1 復(fù)合膜的制備
在2.5M的H2SO4中,經(jīng)不同載波電位鈍化后,304不銹鋼試樣的AFM圖像示于圖2。圖2(a),(b)分別代表 Eh=0.8V(AV800)和 Eh=1.02V(AV1020)試樣。由圖2可知,兩種載波電位條件下形成的氧化膜形態(tài)有明顯的不同,AV1020試樣的表面出現(xiàn)直徑為10~50nm的小孔,而AV800試樣表面是均勻無孔的,這與文獻(xiàn)報(bào)道相一致[7]。

圖2 304不銹鋼在不同載波鈍化電位(Eh)后的AFM圖(a)AV800;(b)AV1020Fig.2 AFM maps of 304stainless steel in 2.5MH2SO4solution at different Ehafter alternating voltage passivation for 10min(a)AV800;(b)AV1020
圖3示出在三種不同基體上采用恒電流法合成PTH膜的電位-時(shí)間關(guān)系曲線。與在惰性電極Pt及未經(jīng)處理的304不銹鋼上合成PTH膜的曲線不同,在AV1020試樣(簡寫為304SS/AV1020)上的電沉積曲線出現(xiàn)兩個(gè)峰,峰值電位分別為0.868V(A)與0.876V(B)。與Schultze等[8]在規(guī)則多孔硅表面電沉積PTH膜的E-t曲線相比較,可推測A段為孔內(nèi)成核區(qū),AB段為孔內(nèi)生長,B點(diǎn)之后是孔外生長。由于304SS/AV1020鈍化膜表面的小孔并不規(guī)則,所得兩峰都是慢峰,沒有明顯的孔內(nèi)與孔外生長的分界點(diǎn)。因此,本實(shí)驗(yàn)只考察了B點(diǎn)以后的復(fù)合膜電化學(xué)性能。
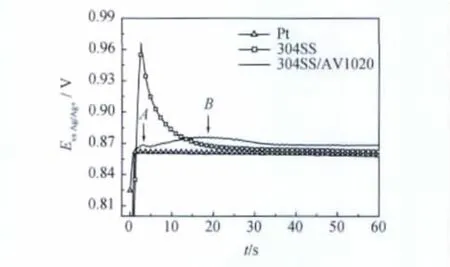
圖3 不同電極上電沉積PTH膜的電位-時(shí)間關(guān)系曲線Fig.3 E-t curves of electrodeposition of PTH film on different samples in 5mM bithiophene+0.1M TBAPF6acetonitrile solution
為考察氧化膜形態(tài)對(duì)電沉積PTH膜的影響,在不同載波條件下形成的304不銹鋼氧化膜上用恒電流法合成PTH膜的電位-時(shí)間關(guān)系曲線示于圖4。由圖4可知,在反應(yīng)40s之后,兩條曲線均表現(xiàn)出聚合電位隨時(shí)間趨于穩(wěn)定,最后恒定在單體的氧化電位約0.85V。這說明在兩種氧化膜試樣表面都有PTH膜生成。與在304SS/AV1020氧化膜上沉積的曲線不同,304SS/AV800氧化膜上沉積PTH膜的合成曲線在沉積之初出現(xiàn)一個(gè)小的電位階躍后,電位便趨于穩(wěn)定,這一點(diǎn)與在Pt電極上有些類似。這是由于304SS/AV800氧化膜致密且陽極溶解速率小。上述兩條氧化膜合成曲線的比較表明,不同形態(tài)的氧化膜對(duì)PTH膜的電沉積過程有很大的影響。
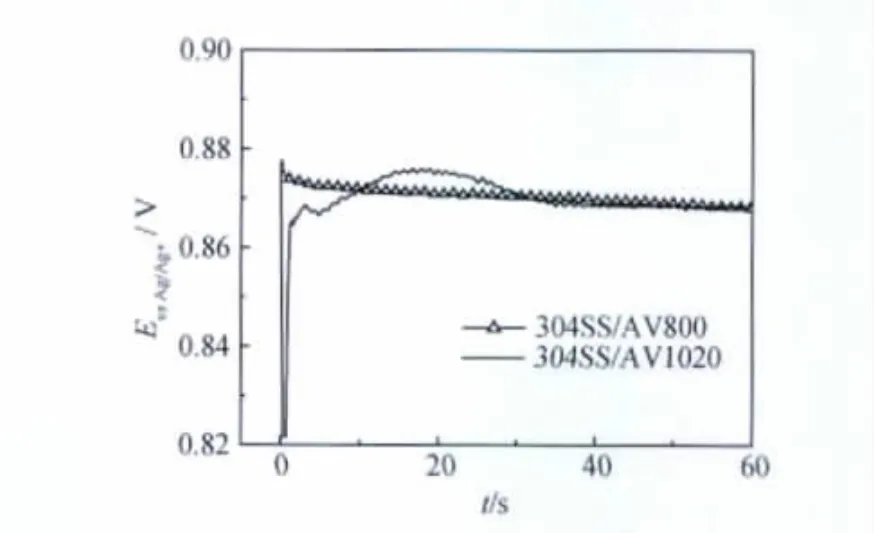
圖4 不同的304不銹鋼載波鈍化膜上電沉積PTH膜的電位-時(shí)間關(guān)系曲線Fig.4 E-t curves of electrodeposition of PTH film on 304SS/AV800and 304SS/AV1020samples in 5mM bithiophene+0.1MTBAPF6acetonitrile solution
2.2 復(fù)合膜的表征
圖5示出兩種載波條件下,合成電量為20mC/cm2的復(fù)合膜在0.1M的TBAPF6乙腈溶液中的循環(huán)伏安曲線。與在相同條件下Pt電極上合成的PTH膜相比,304SS/AV 1020/PTH 與304SS/AV 800/PTH 復(fù)合膜的循環(huán)伏安曲線幾乎重疊,且出現(xiàn)一個(gè)氧化峰,兩個(gè)還原峰。氧化峰電位為Epa=0.695V,兩個(gè)還原峰電位分別為Epc1=0.642V,Epc2=0.305V。僅在半峰寬上略有不同,依次為0.068,0.057V和0.060V。這說明在金屬氧化物上生成的PTH層的電化學(xué)氧化還原性能與Pt惰性電極相一致。
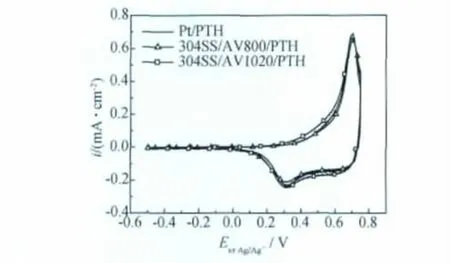
圖5 不同載波條件制備的復(fù)合膜在0.1MTBAPF6乙腈溶液中的循環(huán)伏安曲線Fig.5 Cyclic voltammograms of different electrodes in monomer-free 0.1MTBAPF6acetonitrile solution
電化學(xué)合成的304SS/AV1020/PTH復(fù)合膜的全反射FTIR譜示于圖6。曲線(c)在500~1500cm-1范圍內(nèi),出現(xiàn)1330,1200,1120,1025cm-1的四個(gè)吸收帶為摻雜態(tài)特征峰。另外,789cm-1為2,5-取代的β位的C—H面外彎曲振動(dòng),690cm-1為2取代的C—H面外彎曲振動(dòng)。由于PTH膜比較薄,因此復(fù)合膜還原態(tài)的紅外光譜曲線(b)在1000~1500cm-1范圍內(nèi),呈現(xiàn)出部分AV1020氧化膜的紅外光譜峰,但在789cm-1處出現(xiàn)的聚噻吩2,5-取代特征峰,驗(yàn)證了304不銹鋼氧化膜表面上包覆有按α-α′規(guī)則連接的PTH膜[9]。
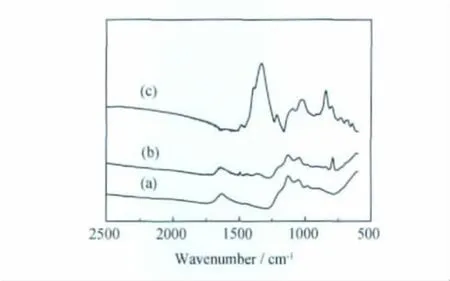
圖6 復(fù)合膜的全反射紅外光譜分析(a)304SS/AV1020氧化膜;(b)304SS/AV800/PTH 復(fù)合膜還原態(tài);(c)304SS/AV1020/PTH 復(fù)合氧化膜Fig.6 FTIR analysis of composite films a)304SS/AV1020film;(b)reductive state of 304SS/AV800/PTH film;(c)oxidation state of 304SS/AV1020/PTH film
PTH作為導(dǎo)電聚合物,電導(dǎo)率是衡量其性能的重要指標(biāo)。未經(jīng)摻雜的無缺陷共軛體系的PTH并不具有導(dǎo)電性。經(jīng)摻雜后,PTH膜才具有一定的導(dǎo)電性。本實(shí)驗(yàn)條件下生成的PTH復(fù)合膜的電導(dǎo)率在5~10S/cm的范圍內(nèi),具有較好的導(dǎo)電性能,屬于導(dǎo)體范疇。
參照ASTM—F1044—05進(jìn)行界面結(jié)合強(qiáng)度的測試,不同條件下試樣的結(jié)合強(qiáng)度列于表1。由表1數(shù)據(jù)可知,經(jīng)載波氧化處理后,PTH膜與基體的結(jié)合力都有很大程度的提高。其中,以304SS/AV1020/PTH試樣的界面結(jié)合強(qiáng)度最大。這是由于304SS/AV1020氧化膜的多孔性,PTH膜在電沉積時(shí)形成了與多孔氧化物的嵌合層[10],提高了PTH膜與基體的界面結(jié)合強(qiáng)度。

表1 PTH膜與基體的結(jié)合強(qiáng)度Table 1 Adhesive strength of PTH film to the substrates
2.3 復(fù)合膜的耐蝕性
304SS/AV800/PTH,304SS/AV1020/PTH 復(fù)合膜與其基底在1MH2SO4溶液中的腐蝕電位-時(shí)間曲線示于圖7。304SS/AV800與304SS/AV1020的初始電位分別為0.357V與0.409V,經(jīng)10h后,電位急驟下降,并隨著時(shí)間的延長基本穩(wěn)定在-0.4V,處于活性溶解狀態(tài)。這說明未經(jīng)后處理的不銹鋼載波氧化膜不能長時(shí)間地保護(hù)304不銹鋼基體[11]。304SS/AV800/PTH 與 304SS/AV1020/PTH 試 樣的初始電位分別為0.677V和0.671V,隨著浸泡時(shí)間的延長開路電位徐徐下降,10h后,電位進(jìn)入平臺(tái)區(qū),表明復(fù)合膜層起到有效的屏障作用。經(jīng)120h的浸泡,復(fù)合膜的開路電位保持在0.4V左右,說明在強(qiáng)酸性介質(zhì)中,包覆PTH復(fù)合膜的不銹鋼仍在鈍化區(qū)內(nèi),復(fù)合膜沒有破損,仍有很強(qiáng)的保護(hù)作用。需要指出的是,浸泡120h后的304SS/AV800/PTH試樣上PTH膜,取出后一擦拭即脫落,而304SS/AV1020/PTH試樣上的PTH膜致密而牢固,取出后擦拭不掉。
圖8示出304SS/AV800/PTH,304SS/AV1020/PTH復(fù)合膜與其基底在1MH2SO4溶液中的陽極極化曲線。由圖8可知,兩種復(fù)合膜的腐蝕電位分別為0.467V與0.579V,較304SS的分別提高約0.8V和0.9V。304SS/AV800/PTH與304SS/AV1020/PTH試樣在極化開始就進(jìn)入鈍化區(qū)。在陽極極化電位0.6V處,兩個(gè)試樣與304SS的電流密度分別為0.73,0.037,23μA/cm2,這說明復(fù)合膜抑制了陽極活性溶解,對(duì)304不銹鋼基體起到有效的保護(hù)作用。304SS/AV800基底的極化電流密度小于304SS/AV1020試樣,這表明其耐蝕性優(yōu)于304SS/AV1020,這與其表面氧化膜致密無孔相一致[12]。然而,304SS/AV1020/PTH卻比304SS/AV800/PTH試樣的過鈍化電位正移約0.2V,維鈍電流密度降低1個(gè)數(shù)量級(jí)左右。這一現(xiàn)象是由于304SS/AV1020氧化膜的多孔性,與PTH形成界面結(jié)合強(qiáng)度高的高分子氧化物嵌合層,提高了膜的耐蝕性。
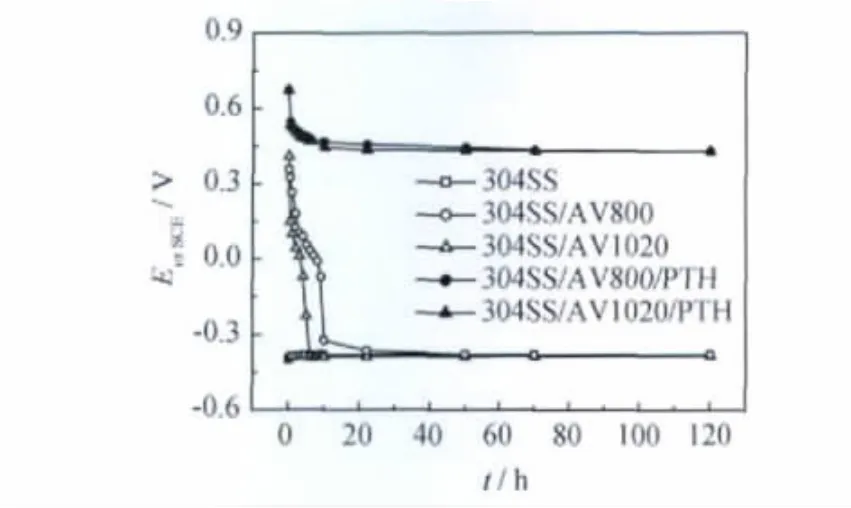
圖7 室溫1MH2SO4溶液中304SS,304SS/AV800,304SS/AV1020與兩復(fù)合膜的腐蝕電位-時(shí)間關(guān)系曲線Fig.7 The Eocp-t curves of 304SS,304SS/AV800,304SS/AV1020and two complex films in 1MH2SO4 solution at room temperature
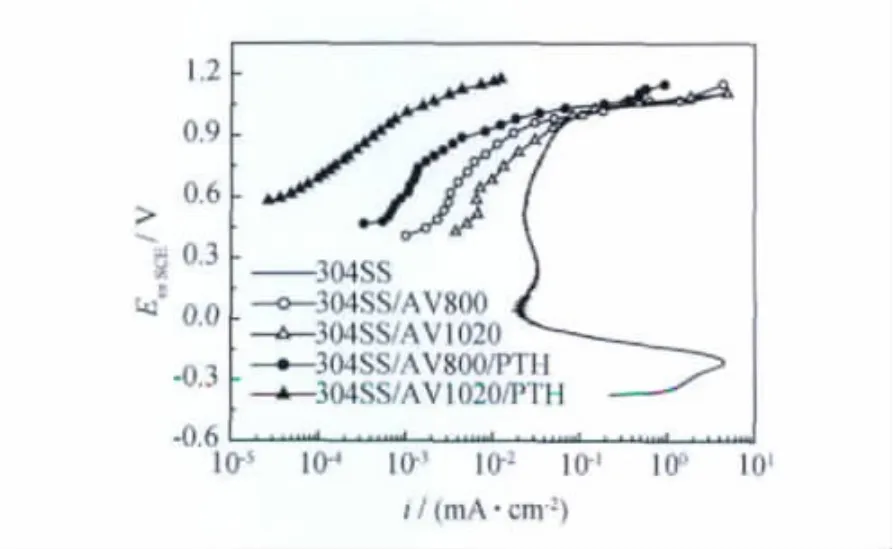
圖8 復(fù)合膜與304SS,304SS/AV800,304SS/AV1020試樣在1MH2SO4中的陽極極化曲線Fig.8 The anodic polarization curves recorded for 304SS,304SS/AV800,304SS/AV1020,304SS/AV800/PTH and 4SS/AV1020/PTH in 1MH2SO4solution at room temperature
為進(jìn)一步考察復(fù)合膜對(duì)金屬基體的保護(hù)作用,在硫酸溶液中考察了浸泡時(shí)間對(duì)交流阻抗譜的影響。304SS/AV1020/PTH試樣在1MH2SO4溶液中浸泡不同時(shí)間的EIS譜示于圖9。由圖9可知,不同浸泡時(shí)間對(duì)應(yīng)的交流阻抗譜的變化趨勢(shì)一致。圖9(a)的復(fù)合膜在曲線的高頻端都表現(xiàn)為一個(gè)容抗弧,代表典型的膜/溶液或金屬/膜界面電容Cdl與反應(yīng)電阻Rct并聯(lián)電路;低頻端則出現(xiàn)電容的行為,這與有機(jī)介質(zhì)中的Nyquist圖相近。選用R(QR)(Q(RW))等效電路進(jìn)行擬合[13]。擬合的304SS/AV1020/PTH 試樣在1M H2SO4溶液中不同浸泡時(shí)間對(duì)應(yīng)的交流阻抗參數(shù)示于表2。隨著浸泡時(shí)間延長,Nyquist圖高頻端的容抗弧半徑增大,反應(yīng)電阻Rct由37.2Ω·cm2增大至118.6Ω·cm2,其原因是PTH膜隨著浸泡時(shí)間延長發(fā)生脫摻雜反應(yīng),膜還原時(shí),其導(dǎo)電性下降,導(dǎo)致反應(yīng)阻力增加;而對(duì)應(yīng)Bode圖中(圖9(b))的相角向低頻端移動(dòng)。浸泡20h后,在頻率1000~100Hz范圍內(nèi),相角有升高的現(xiàn)象,這是因?yàn)镻TH膜還原時(shí),膜會(huì)縮水,減小表面的微孔[14],對(duì)應(yīng)極限電容YA呈小幅度的減小。經(jīng)120h的浸泡,低頻端相角值仍很高,表現(xiàn)為電容行為,說明復(fù)合膜很完整,沒有破損,此時(shí)的開路電位處在0.4V左右,表明不銹鋼基體仍維持在鈍化狀態(tài)。
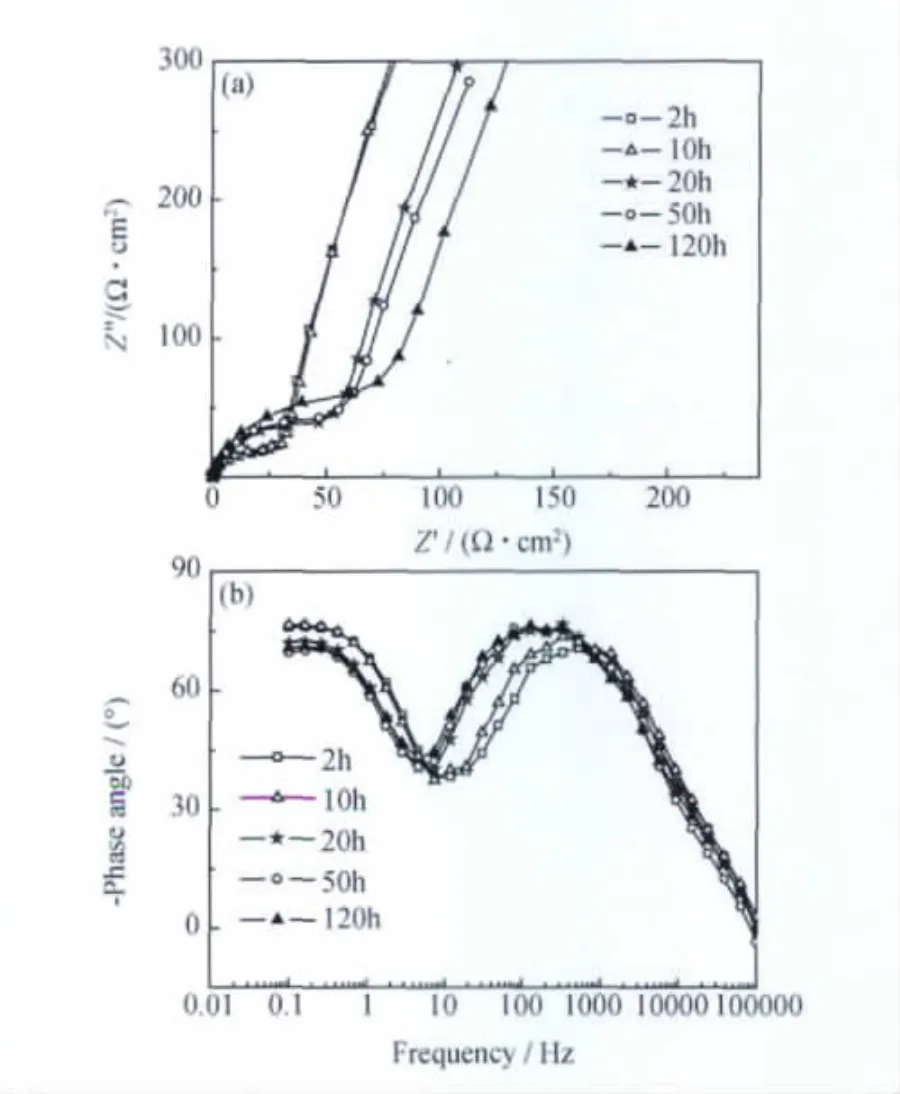
圖9 304SS/AV1020/PTH試樣在1MH2SO4溶液中浸泡不同時(shí)間的EIS譜 (a)Nyquist圖;(b)Bode圖Fig.9 EIS curves for 304SS/AV1020/PTH sample in 1MH2SO4solution after various exposure time at room temperature (a)Nyquist;(b)Bode
3 結(jié)論
(1)以304不銹鋼為基體,通過載波鈍化后在其氧化膜上電沉積PTH膜,制備了304SS/氧化膜/PTH的復(fù)合膜。
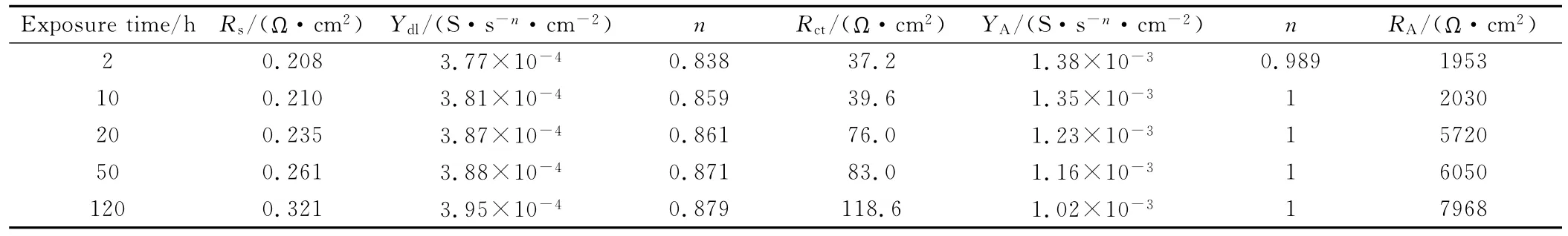
表2 304SS/AV1020/PTH試樣在1MH2SO4溶液中不同浸泡時(shí)間對(duì)應(yīng)的交流阻抗參數(shù)Table 2 Parameters of the EIS diagram for 304SS/AV1020/PTH sample in 1MH2SO4solution after various exposure time
(2)通過紅外光譜與循環(huán)伏安法表征了復(fù)合膜的分子結(jié)構(gòu)、表面形態(tài)與電化學(xué)性能。結(jié)果表明,復(fù)合膜中的PTH層的分子結(jié)構(gòu)按α-α′規(guī)則連接,并具有良好的電化學(xué)氧化還原可逆性。復(fù)合膜的電導(dǎo)率在5~10S/cm的范圍內(nèi)。
(3)304SS/PTH 和304SS/AV800/PTH,304SS/AV1020/PTH復(fù)合膜的界面結(jié)合強(qiáng)度分別為0.81,4.98MPa和5.10MPa。在1MH2SO4溶液中的耐蝕性測試結(jié)果,兩復(fù)合膜的腐蝕電位正移0.80V,使304不銹鋼基體保持在鈍化狀態(tài),抑制了304不銹鋼基體的陽極活性溶解,提高了耐蝕性能。比較而言,由于304SS/AV1020表面多孔性與PTH膜形成多孔氧化物的嵌合層,使得304SS/AV1020/PTH復(fù)合膜的電化學(xué)、電學(xué)及界面結(jié)合強(qiáng)度均優(yōu)于304SS/AV800/PTH復(fù)合膜。
[1]HU X,WANG G M,WONG T K S.Effect of aqueous and organic solvent ratio on the electropolymerization of bithiophene in the mixed solutions[J].Synthetic Metals,1999,106(3):145-150.
[2]RONCALI J.Conjugated poly(thiophenes):synthesis,functionalization,and applications[J].Chemical Reviews,1992,92(4):711-738.
[3]REN S,BARKEY D.Electrochemically prepared poly(3-methythiophene)films for passivation of 430stainless steel[J].J Electrochem Soc,1992,139(4):1021-1026.
[4]ZHANG J X,YUAN J,QIAO Y N,et al.The corrosion and passivation of SS304stainless steel under square wave electric field[J].Materials Chemistry and Physics,2003,79(1):43-48.
[5]FUJIMOTO S,TSUJINO K,SHIBATA T.Growth and properties of Cr-rich thick and porous oxide films on type 304stainless steel formed by square wave potential pulse polarization[J].Electrochimica Acta,2001,47(4):543-551.
[6]ASTM standard F1044—05,standard test method for shear testing of calcium phosphate coatings and metallic coatings[S].
[7]張俊喜,喬亦男,曹楚南,等.不銹鋼載波鈍化的研究和應(yīng)用[J].上海電力學(xué)院學(xué)報(bào),2004,20(3):27-34.ZHANG Jun-xi,QIAO Yi-nan,CAO Chu-nan,et al.The investi-gation and application of A.V.passivation of stainless steel[J].Journal of Shanghai University of Electric Power,2004,20(3):27-34.
[8]SCHULTZE J W,JUNG K G.Regular nanostructured systems formed electrochemically:deposition of electroactive polybithiophene into porous silicon[J].Electrochimica Acta,1995,40(10):1369-1383.
[9]MOREA G,LSABBATINI L,ZAMBONIN P G,et al.Modification of polybithiophene by electrochemical cycling studied by ToF-SIMS and XPS[J].Macromolecules,1991,24(12):3630-3637.
[10]金緒剛,黃承亞,龔克成.本征導(dǎo)電聚合物涂層及界面[J].化學(xué)研究與應(yīng)用,1997,9(4):332-337.JIN Xu-gang,HUANG Cheng-ya,GONG Ke-cheng.Intrinsically conducting polymer coatings and interfaces[J].Chemical Research and Application,1997,9(4):332-337.
[11]KWIATKOWSKI L,MANSFELD F.Surface modification of stainless steel by an alternating voltage process[J].J Electrochem Soc,1993,140(3):39-41.
[12]張俊喜,顏立成,魏增福,等.不銹鋼載波鈍化膜的生長過程[J].金屬學(xué)報(bào),2004,40(4):404-410.ZHANG Jun-xi,YAN Li-cheng,WEI Zeng-fu,et al.Formation of passive film on 304stainless steel under alternating voltage electric field[J].Acta Metallurgica Sinica,2004,40(4):404-410.
[13]梁成浩,陳婉,黃乃寶,等.惰性電極上電合成聚噻吩膜的分形特征研究[J].材料科學(xué)與工藝,2009,17(1):8-12.LIANG Cheng-h(huán)ao,CHEN Wan,HUANG Nai-bao,et al.Fractal characteristics of electrochemical synthetic polybithiophene on inert electrode[J].Materials Science and Technology,2009,17(1):8-12.
[14]WESSLING B,POSDORFER J.Corrosion prevention with an organic metal(polyaniline):corrosion test results[J].Electro-chimica Acta,1999,44(12):2139-2147.
猜你喜歡
趣味(數(shù)學(xué))(2022年3期)2022-06-02 02:32:52
山東冶金(2022年1期)2022-04-19 13:40:20
小哥白尼(軍事科學(xué))(2021年12期)2021-03-29 00:49:18
山東冶金(2019年1期)2019-03-30 01:35:32
中國特種設(shè)備安全(2018年10期)2018-12-18 02:17:18
酒·飲料技術(shù)裝備(2018年1期)2018-04-28 09:09:10
中學(xué)生數(shù)理化·八年級(jí)物理人教版(2017年10期)2018-01-22 03:04:00
制造技術(shù)與機(jī)床(2017年8期)2017-11-27 02:10:21
商洛學(xué)院學(xué)報(bào)(2017年2期)2017-05-17 05:19:50
石油化工建設(shè)(2016年4期)2016-02-27 15:03:16
